Station d'Ecologie Théorique et Expérimentale de Moulis, CNRS, Moulis, France.
Département de Biochimie, Centre Robert-Cedergren, Université de Montréal, Montréal, Québec, Canada.
BMC Evol Biol. 2019 Jan 11;19(1):21. doi: 10.1186/s12862-019-1350-2.
Multiple Sequence Alignments (MSAs) are the starting point of molecular evolutionary analyses. Errors in MSAs generate a non-historical signal that can lead to incorrect inferences. Therefore, numerous efforts have been made to reduce the impact of alignment errors, by improving alignment algorithms and by developing methods to filter out poorly aligned regions. However, MSAs do not only contain alignment errors, but also primary sequence errors. Such errors may originate from sequencing errors, from assembly errors, or from erroneous structural annotations (such as incorrect intron/exon boundaries). Even though their existence is acknowledged, the impact of primary sequence errors on evolutionary inference is poorly characterized.
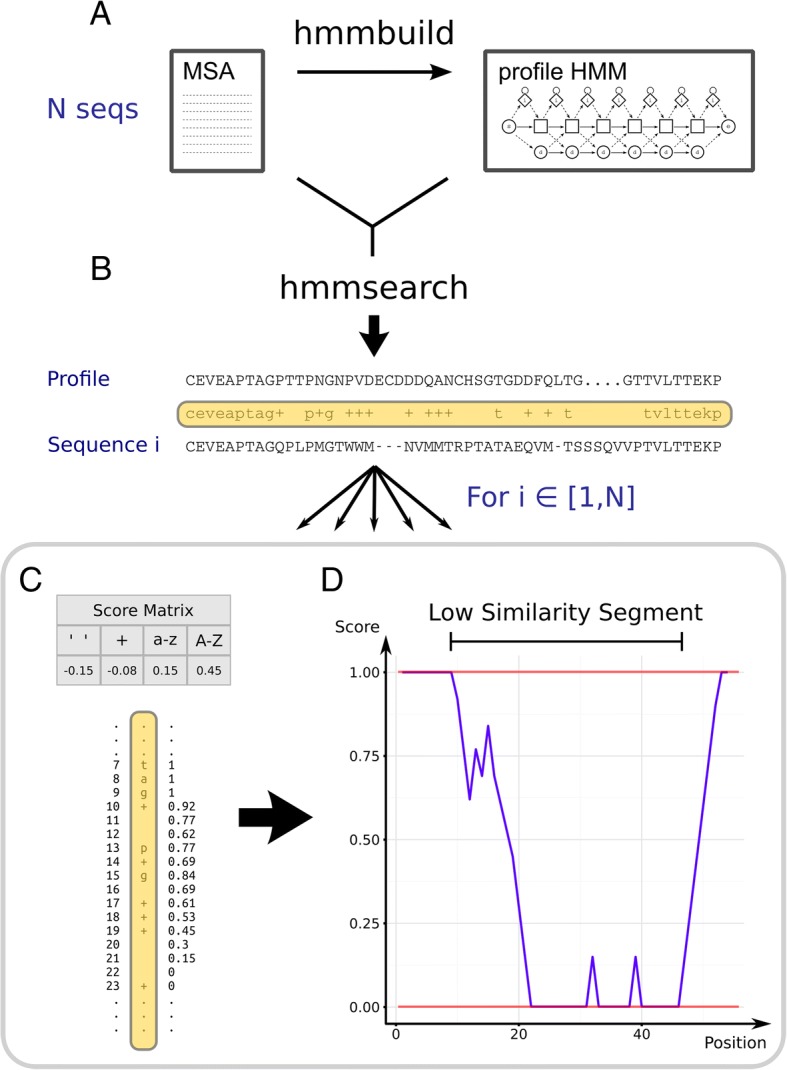
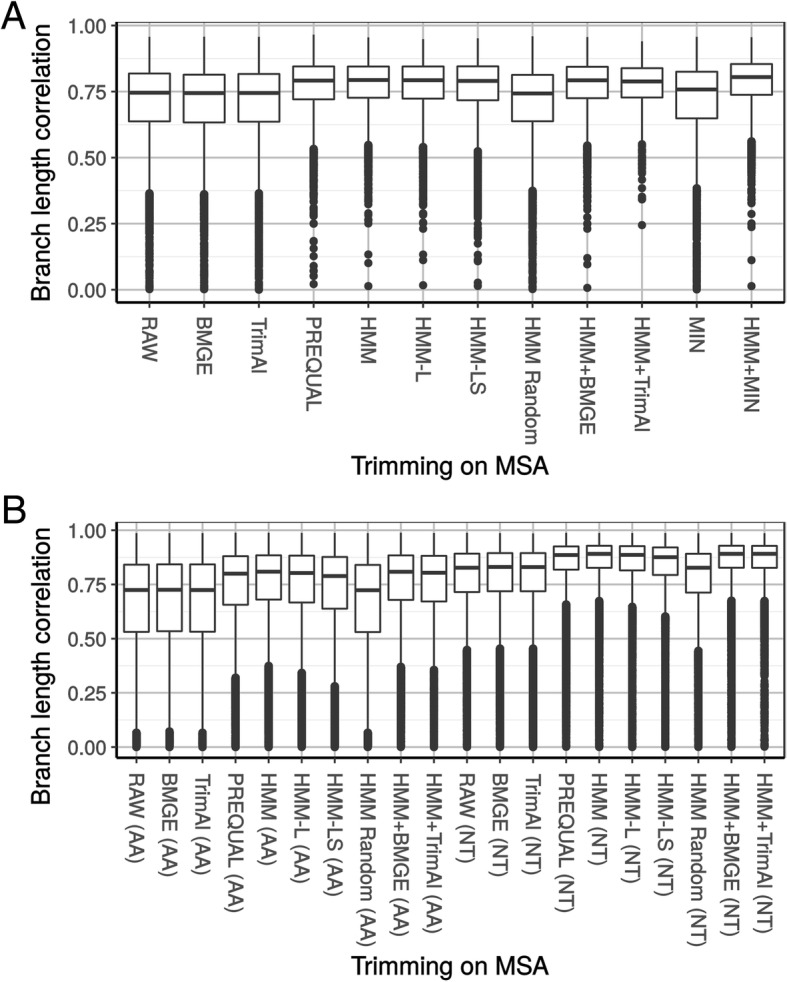
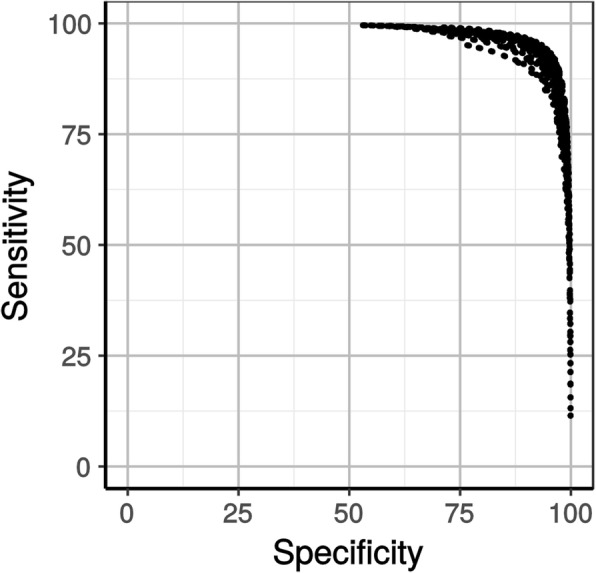
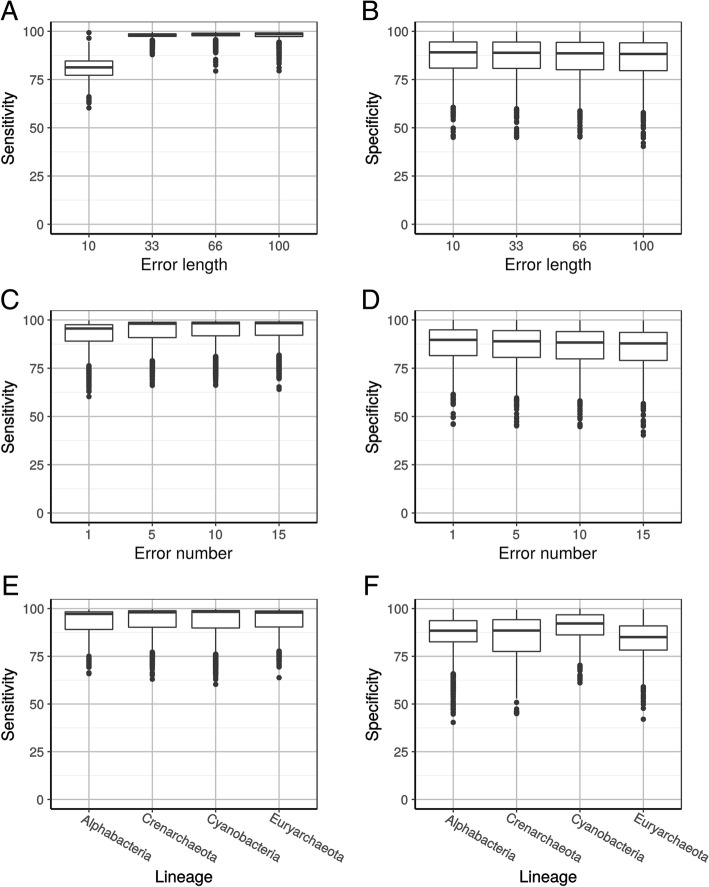
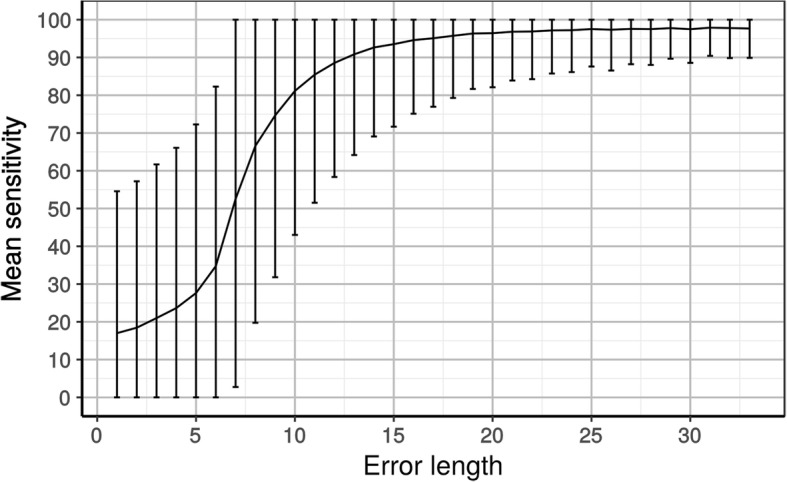
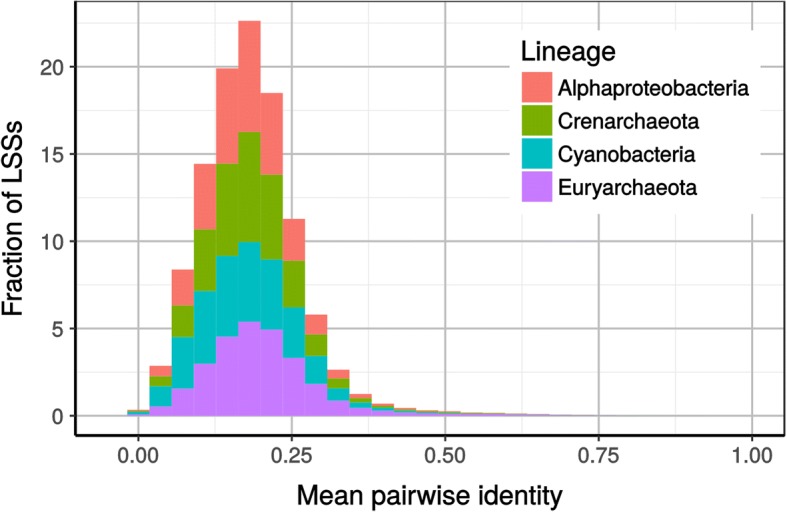
In a first step to fill this gap, we have developed a program called HmmCleaner, which detects and eliminates these errors from MSAs. It uses profile hidden Markov models (pHMM) to identify sequence segments that poorly fit their MSA and selectively removes them. We assessed its performances using > 700 amino-acid MSAs from prokaryotes and eukaryotes, in which we introduced several types of simulated primary sequence errors. The sensitivity of HmmCleaner towards simulated primary sequence errors was > 95%. In a second step, we compared the impact of segment filtering software (HmmCleaner and PREQUAL) relative to commonly used block-filtering software (BMGE and TrimAI) on evolutionary analyses. Using real data from vertebrates, we observed that segment-filtering methods improve the quality of evolutionary inference more than the currently used block-filtering methods. The formers were especially effective at improving branch length inferences, and at reducing false positive rate during detection of positive selection.
Segment filtering methods such as HmmCleaner accurately detect simulated primary sequence errors. Our results suggest that these errors are more detrimental than alignment errors. However, they also show that stochastic (sampling) error is predominant in single-gene evolutionary inferences. Therefore, we argue that MSA filtering should focus on segment instead of block removal and that more studies are required to find the optimal balance between accuracy improvement and stochastic error increase brought by data removal.
多序列比对(MSAs)是分子进化分析的起点。比对中的错误会产生非历史信号,从而导致错误的推断。因此,人们已经做出了许多努力来减少对齐错误的影响,例如改进对齐算法和开发过滤不良对齐区域的方法。然而,MSAs 不仅包含对齐错误,还包含原始序列错误。这些错误可能源于测序错误、组装错误或错误的结构注释(例如不正确的内含子/外显子边界)。尽管已经认识到它们的存在,但原始序列错误对进化推断的影响还没有得到很好的描述。
为了填补这一空白,我们开发了一个名为 HmmCleaner 的程序,它可以从 MSAs 中检测和消除这些错误。它使用轮廓隐马尔可夫模型(pHMM)来识别与 MSAs 拟合不佳的序列段,并选择性地删除它们。我们使用来自原核生物和真核生物的 >700 个氨基酸 MSAs 评估了它的性能,在这些 MSAs 中引入了几种类型的模拟原始序列错误。HmmCleaner 对模拟原始序列错误的敏感性 >95%。在第二步中,我们比较了片段过滤软件(HmmCleaner 和 PREQUAL)与常用的块过滤软件(BMGE 和 TrimAI)对进化分析的影响。使用来自脊椎动物的真实数据,我们观察到片段过滤方法比当前使用的块过滤方法更能提高进化推断的质量。前者在改进分支长度推断和减少正选择检测中的假阳性率方面尤其有效。
片段过滤方法(如 HmmCleaner)可以准确检测模拟的原始序列错误。我们的结果表明,这些错误比对齐错误更具危害性。然而,它们也表明,在单基因进化推断中,随机(抽样)错误占主导地位。因此,我们认为 MSA 过滤应该侧重于片段而不是块的去除,并且需要更多的研究来找到在准确性提高和数据去除带来的随机误差增加之间的最佳平衡。