Marine Biological Association of the United Kingdom, The Laboratory, Citadel Hill, Plymouth PL1 2PBDevon, UK.
BMC Bioinformatics. 2012 May 30;13:117. doi: 10.1186/1471-2105-13-117.
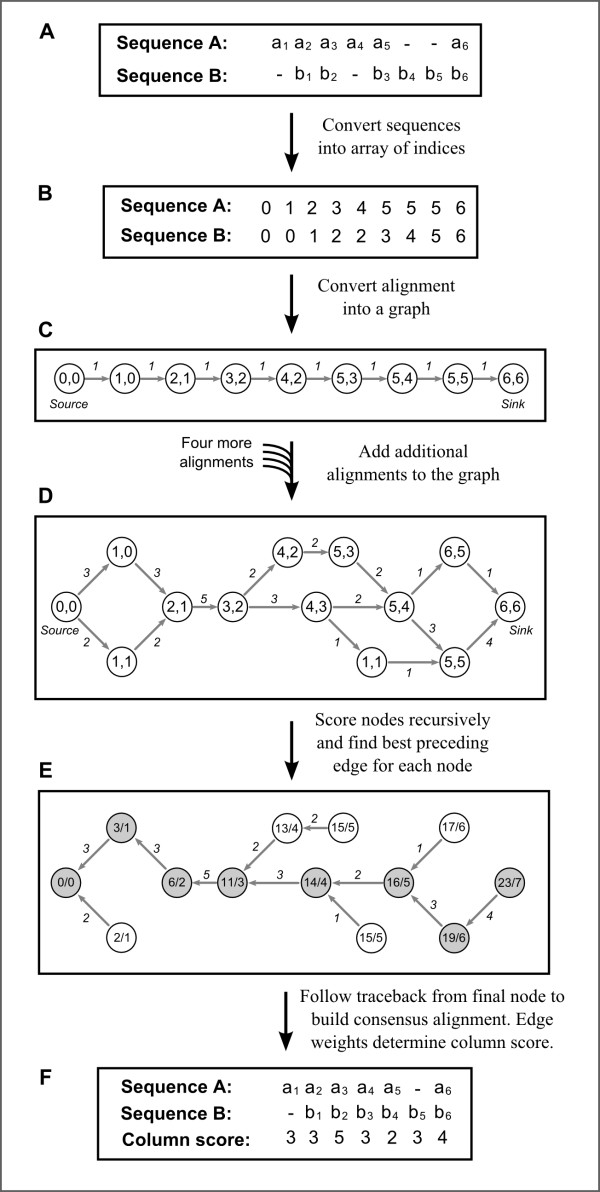
The generation of multiple sequence alignments (MSAs) is a crucial step for many bioinformatic analyses. Thus improving MSA accuracy and identifying potential errors in MSAs is important for a wide range of post-genomic research. We present a novel method called MergeAlign which constructs consensus MSAs from multiple independent MSAs and assigns an alignment precision score to each column.
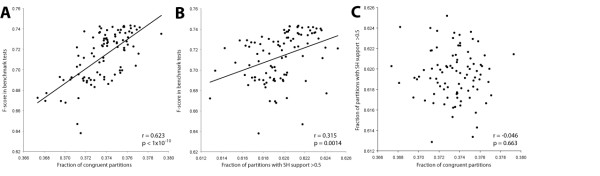
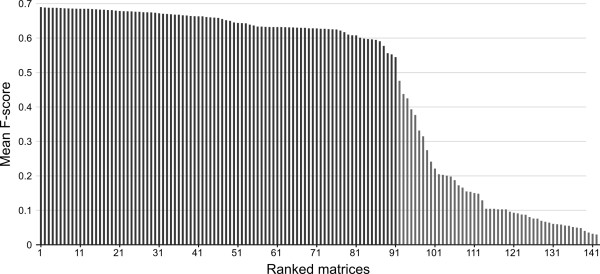
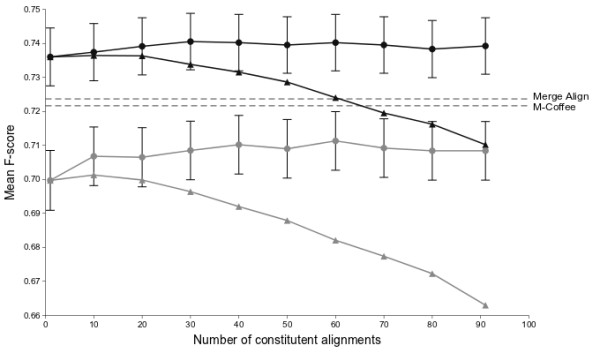
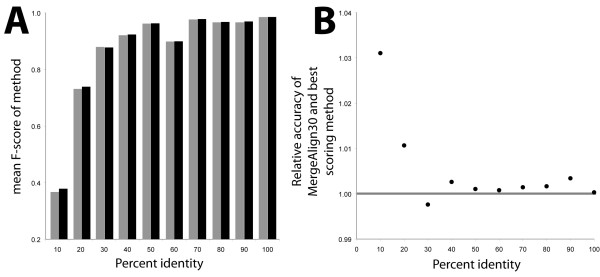
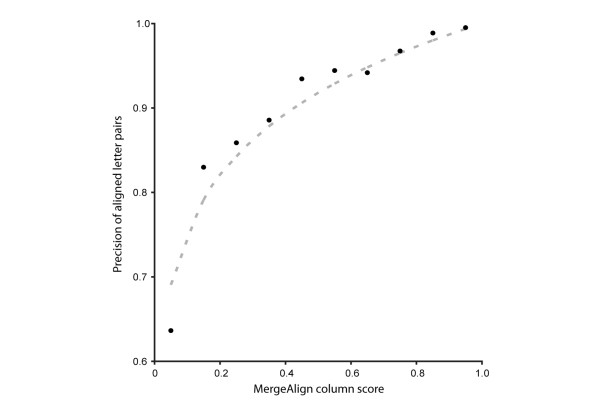
Using conventional benchmark tests we demonstrate that on average MergeAlign MSAs are more accurate than MSAs generated using any single matrix of sequence substitution. We show that MergeAlign column scores are related to alignment precision and hence provide an ab initio method of estimating alignment precision in the absence of curated reference MSAs. Using two novel and independent alignment performance tests that utilise a large set of orthologous gene families we demonstrate that increasing MSA performance leads to an increase in the performance of downstream phylogenetic analyses.
Using multiple tests of alignment performance we demonstrate that this novel method has broad general application in biological research.
多序列比对(MSA)的生成是许多生物信息学分析的关键步骤。因此,提高 MSA 的准确性并识别 MSA 中的潜在错误对于广泛的后基因组研究非常重要。我们提出了一种称为 MergeAlign 的新方法,该方法可以从多个独立的 MSA 构建共识 MSA,并为每个列分配一个对齐精度得分。
使用常规基准测试,我们证明平均而言,MergeAlign MSA 比使用任何单个序列替换矩阵生成的 MSA 更准确。我们表明,MergeAlign 列得分与对齐精度相关,因此提供了一种在没有经过精心整理的参考 MSA 的情况下估计对齐精度的初始方法。使用两个利用大型直系同源基因家族的新的独立对齐性能测试,我们证明了增加 MSA 性能会导致下游系统发育分析性能的提高。
使用多种对齐性能测试,我们证明了这种新方法在生物研究中有广泛的应用。