Tang Lu, Ma Yan, Liu Xiao-Lu, Chen Lu, Fan Dong-Sheng
Department of Neurology, Peking University Third Hospital, 49 North Garden Road, Haidian District, Beijing, 100191 People's Republic of China.
Transl Neurodegener. 2019 Jan 8;8:2. doi: 10.1186/s40035-018-0142-8. eCollection 2019.
mutations are the most common cause of amyotrophic lateral sclerosis (ALS) in non-Caucasian patients. Detailed natural history profiles of -mutant patients will be beneficial for the strategy and interpretation of future 1-targeted clinical practice.
Mutational distribution, age at onset (AAO), site of onset, diagnostic delay, disease progression (rate of ALSFRS-R decrease, ΔFS) and survival were analysed. Further comparisons between heredity of disease, gender, and mutations were performed.
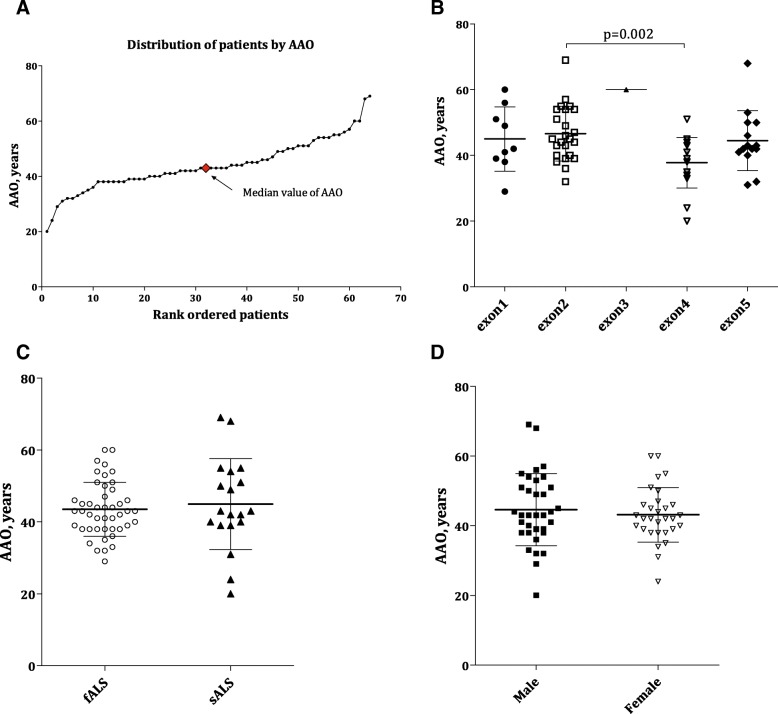
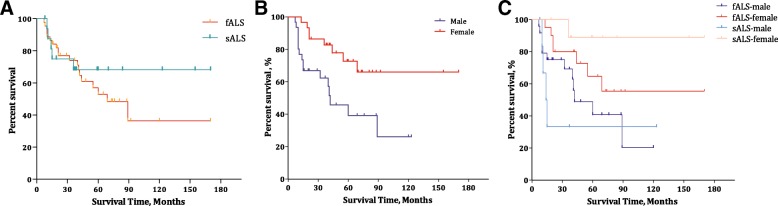
Sixty-six cases with 43 mutations were included and analysed, with p.His47Arg as the leading mutation and seven novel variants identified. The mean (SD) AAO was 43.92 years (9.24) for all subjects, with a significant difference between patients carrying mutations in exon 2 ( = 24,46.83, 8.31) and exon 4 ( = 18, 37.75, 7.67) ( = 0.002). The median (IQR) diagnostic delay from symptom onset was 14.50 (6.00-36.50) months for all -mutant patients, 9.50 (4.75-24.25) months for males and 24.00 (9.50-47.50) months for females, revealing a gender difference ( = 0.009). Similar advantages in median (IQR) ΔFS [male: female, 0.55 (0.24-0.94) vs 0.19 (0.06-0.90), = 0.041] and mean (95% CI) survival [57.4 (38.90-75.90) months vs 125.6 (99.80-151.50) months, = 0.006] were also observed in females, both of which existed in sporadic ALS only when stratified by familiar or sporadic ALS.
The results highlight a distinct mutational distribution and natural history spectrum in ALS patients carrying mutations in China. A prominent mild disease progression was observed in female patients, which had rarely been reported in the previous literature. This finding, together with the detailed analysis of natural history among each mutation, can have important implications for future genetic counselling and -targeted clinical trials.
突变是非白种人肌萎缩侧索硬化症(ALS)最常见的病因。详细了解携带突变患者的自然病史概况将有助于未来针对该靶点的临床实践策略制定和解读。
分析突变分布、发病年龄(AAO)、发病部位、诊断延迟、疾病进展(ALSFRS-R降低率,ΔFS)和生存率。进一步比较疾病遗传、性别和突变之间的差异。
纳入并分析了66例携带43种突变的病例,以p.His47Arg为主要突变,并鉴定出7种新变体。所有受试者的平均(标准差)发病年龄为43.92岁(9.24),外显子2突变患者(n = 24,46.83,8.31)和外显子4突变患者(n = 18,37.75,7.67)之间存在显著差异(P = 0.002)。所有携带突变患者从症状出现到诊断的中位(IQR)延迟为14.50(6.00 - 36.50)个月,男性为9.50(4.75 - 24.25)个月,女性为24.00(9.50 - 47.50)个月,显示出性别差异(P = 0.009)。在女性中,中位(IQR)ΔFS[男性:女性,0.55(0.24 - 0.94)对0.19(0.06 - 0.90),P = 0.041]和平均(95%CI)生存率[57.4(38.90 - 75.90)个月对125.6(99.80 - 151.50)个月,P = 0.006]也有类似优势,仅在按家族性或散发性ALS分层时,两者均存在于散发性ALS中。
结果突出了中国携带该突变的ALS患者独特的突变分布和自然病史谱。女性患者观察到明显的轻度疾病进展,这在以前的文献中很少报道。这一发现,连同对每种突变自然病史的详细分析,可能对未来的遗传咨询和针对该靶点的临床试验具有重要意义。