Minio Andrea, Massonnet Mélanie, Figueroa-Balderas Rosa, Vondras Amanda M, Blanco-Ulate Barbara, Cantu Dario
Department of Viticulture and Enology, University of California Davis, Davis, CA.
Department of Plant Sciences, University of California Davis, Davis, CA.
G3 (Bethesda). 2019 Mar 7;9(3):755-767. doi: 10.1534/g3.118.201008.
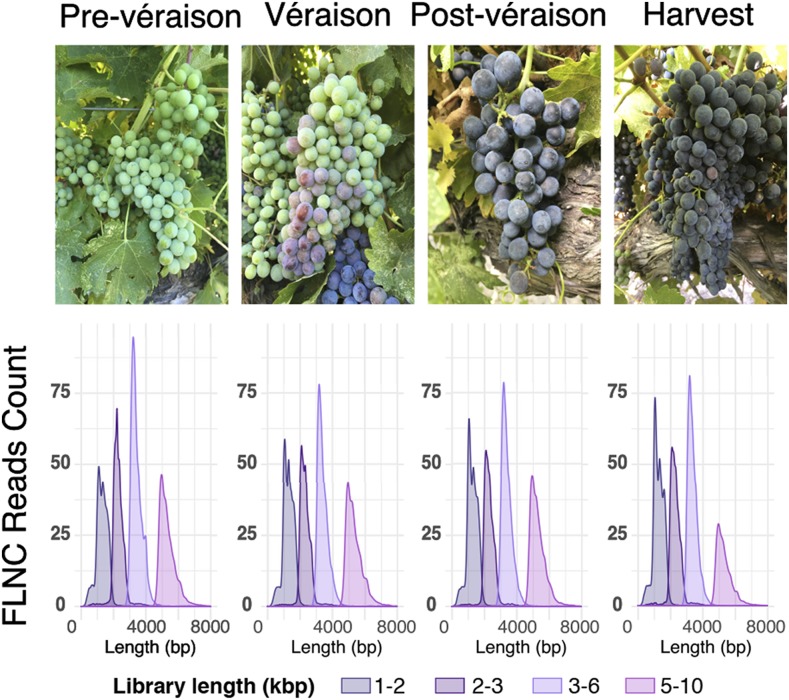
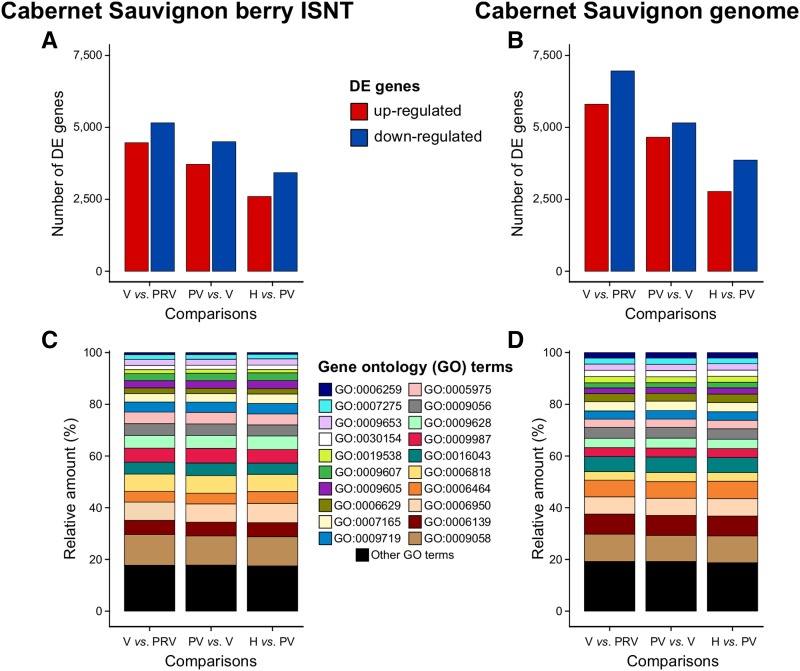
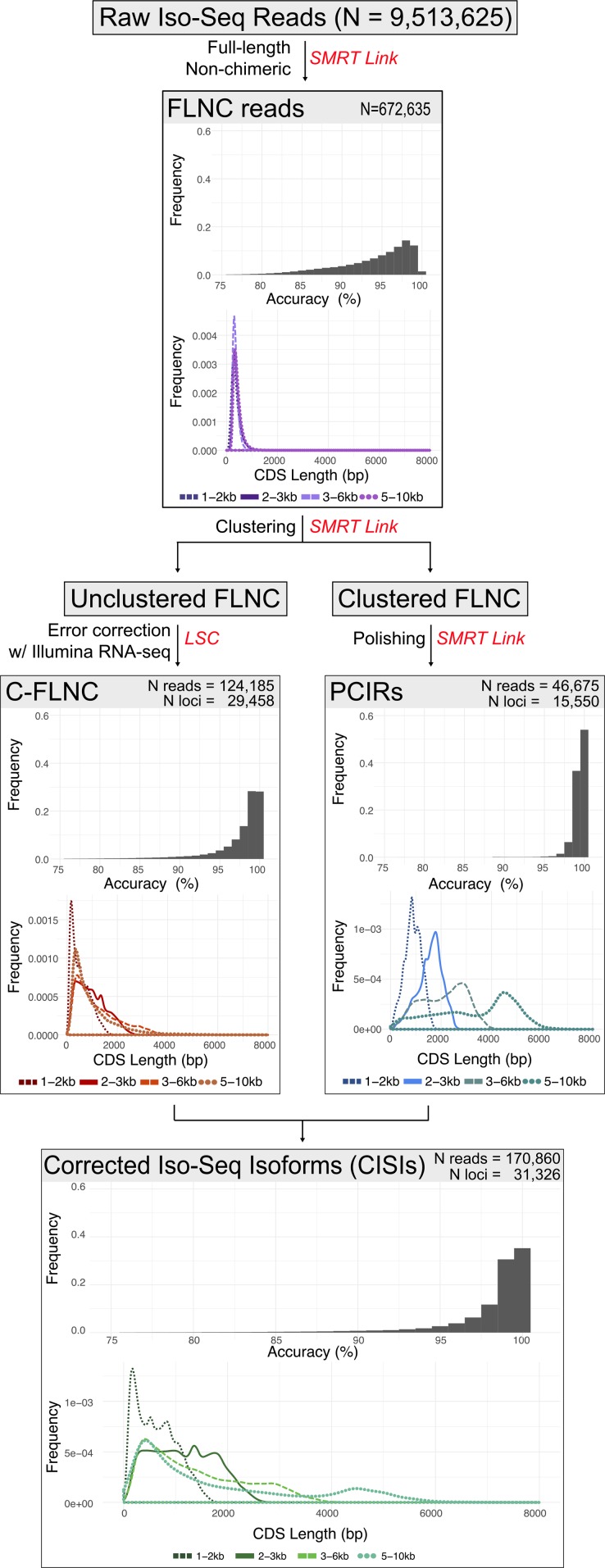
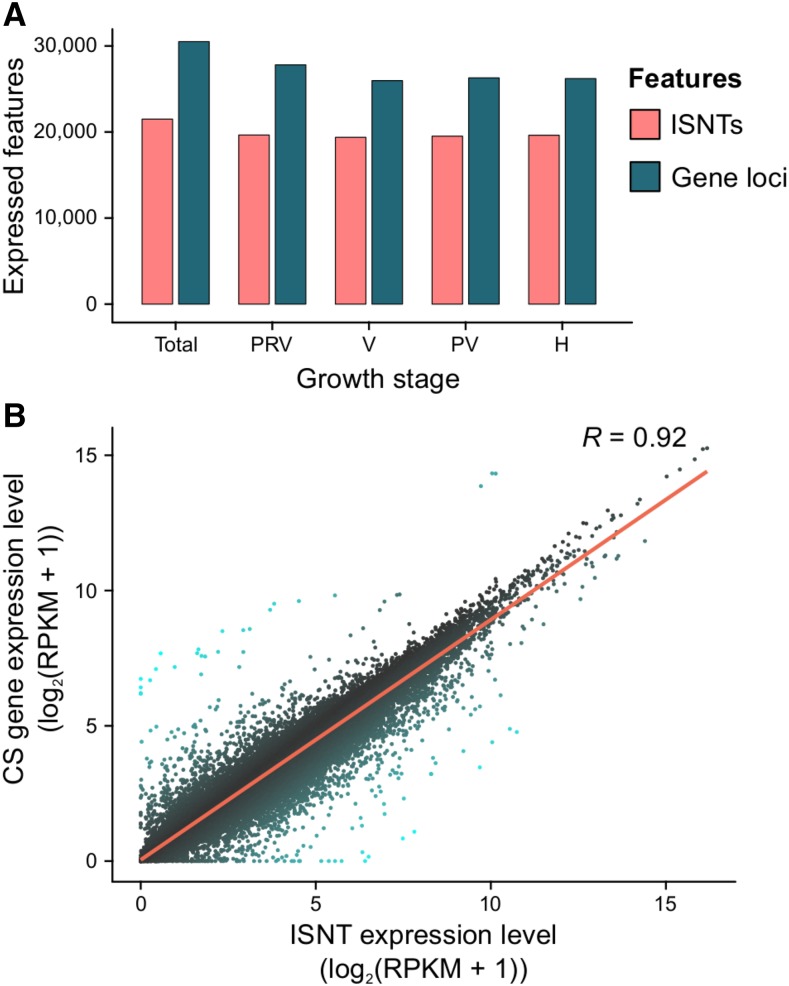
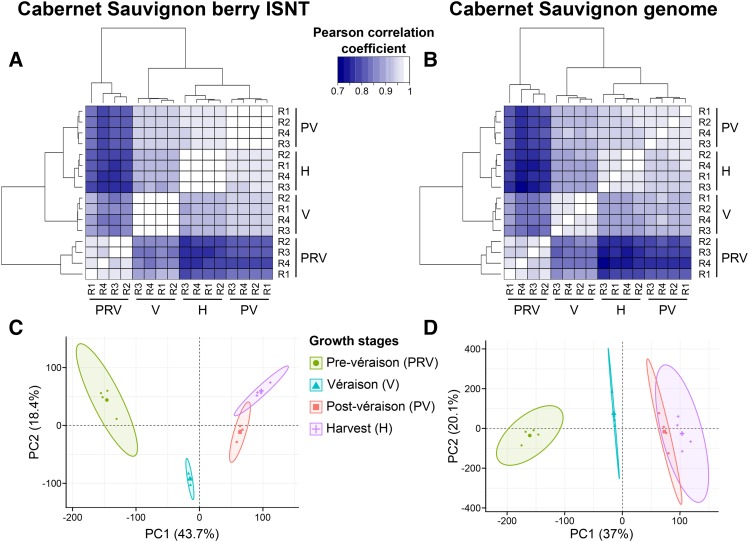
Transcriptomics has been widely applied to study grape berry development. With few exceptions, transcriptomic studies in grape are performed using the available genome sequence, PN40024, as reference. However, differences in gene content among grape accessions, which contribute to phenotypic differences among cultivars, suggest that a single reference genome does not represent the species' entire gene space. Though whole genome assembly and annotation can reveal the relatively unique or "private" gene space of any particular cultivar, transcriptome reconstruction is a more rapid, less costly, and less computationally intensive strategy to accomplish the same goal. In this study, we used single molecule-real time sequencing (SMRT) to sequence full-length cDNA (Iso-Seq) and reconstruct the transcriptome of Cabernet Sauvignon berries during berry ripening. In addition, short reads from ripening berries were used to error-correct low-expression isoforms and to profile isoform expression. By comparing the annotated gene space of Cabernet Sauvignon to other grape cultivars, we demonstrate that the transcriptome reference built with Iso-Seq data represents most of the expressed genes in the grape berries and includes 1,501 cultivar-specific genes. Iso-Seq produced transcriptome profiles similar to those obtained after mapping on a complete genome reference. Together, these results justify the application of Iso-Seq to identify cultivar-specific genes and build a comprehensive reference for transcriptional profiling that circumvents the necessity of a genome reference with its associated costs and computational weight.
转录组学已被广泛应用于研究葡萄果实发育。除少数例外,葡萄的转录组学研究均以现有的基因组序列PN40024作为参考进行。然而,葡萄品种间基因含量的差异导致了品种间的表型差异,这表明单一的参考基因组并不能代表该物种的全部基因空间。虽然全基因组组装和注释可以揭示任何特定品种相对独特或“私有”的基因空间,但转录组重建是实现相同目标的一种更快速、成本更低且计算强度更小的策略。在本研究中,我们使用单分子实时测序(SMRT)对全长cDNA(Iso-Seq)进行测序,并重建赤霞珠葡萄果实成熟过程中的转录组。此外,利用成熟果实的短读长序列对错表达较低的异构体进行纠错,并分析异构体表达情况。通过将赤霞珠的注释基因空间与其他葡萄品种进行比较,我们证明利用Iso-Seq数据构建的转录组参考代表了葡萄果实中大多数表达的基因,其中包括1501个品种特异性基因。Iso-Seq产生的转录组图谱与在完整基因组参考上进行映射后获得的图谱相似。总之,这些结果证明了Iso-Seq在鉴定品种特异性基因和构建转录谱综合参考方面的应用价值,该方法避免了使用基因组参考及其相关成本和计算负担的必要性。