Price Adam, Gibas Cynthia
Department of Bioinformatics and Genomics, University of North Carolina at Charlotte, Charlotte, North Carolina, United States of America.
PLoS One. 2017 Jul 11;12(7):e0180904. doi: 10.1371/journal.pone.0180904. eCollection 2017.
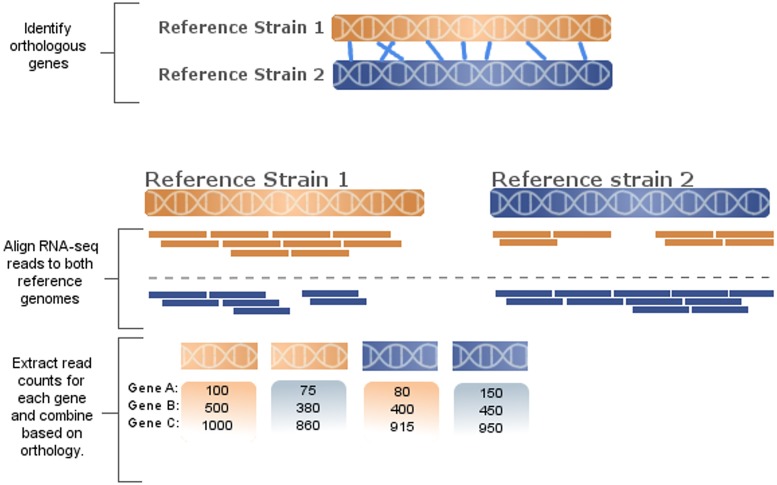
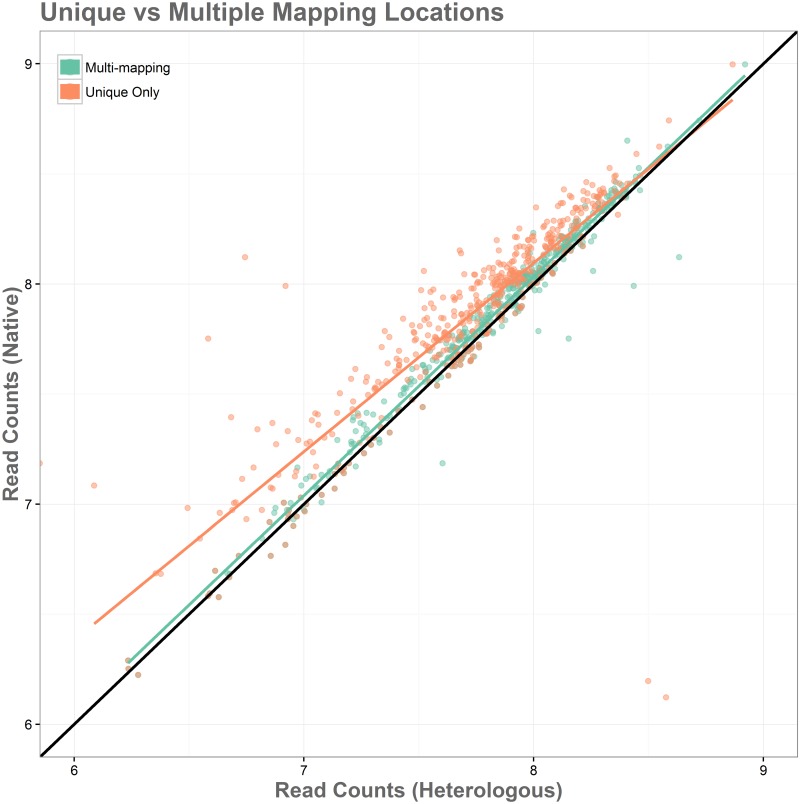
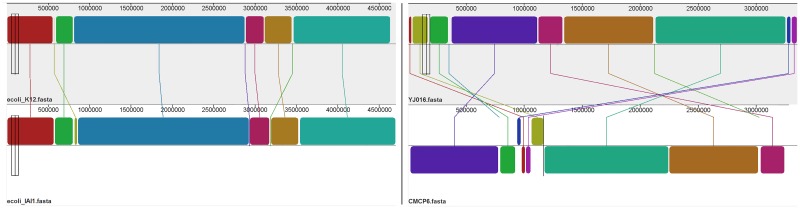
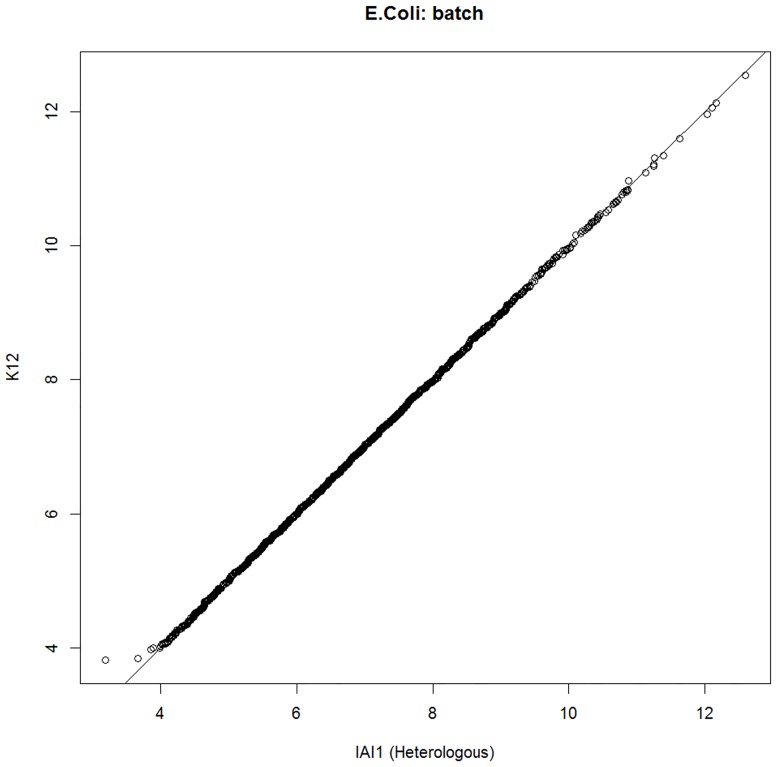
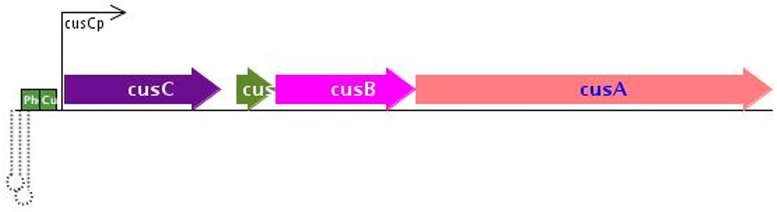
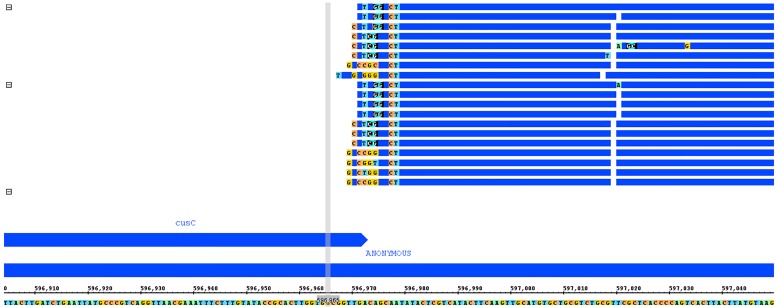
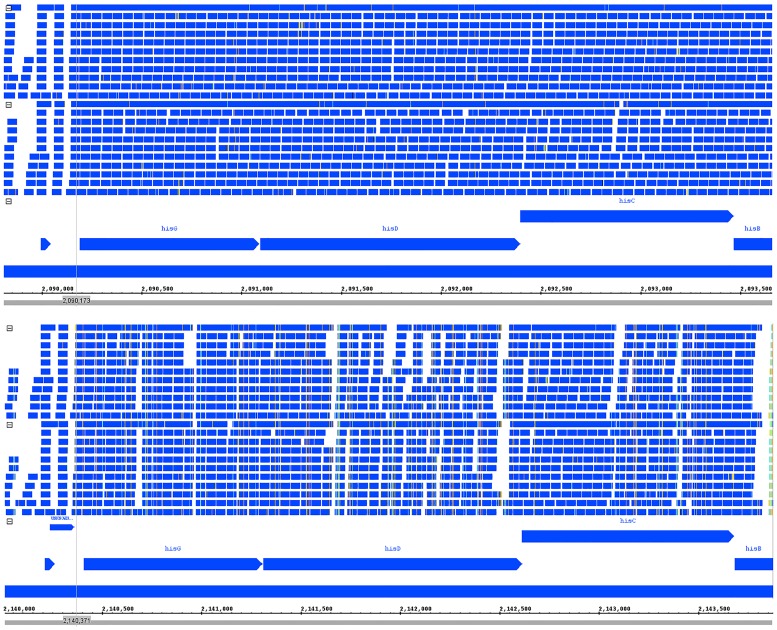
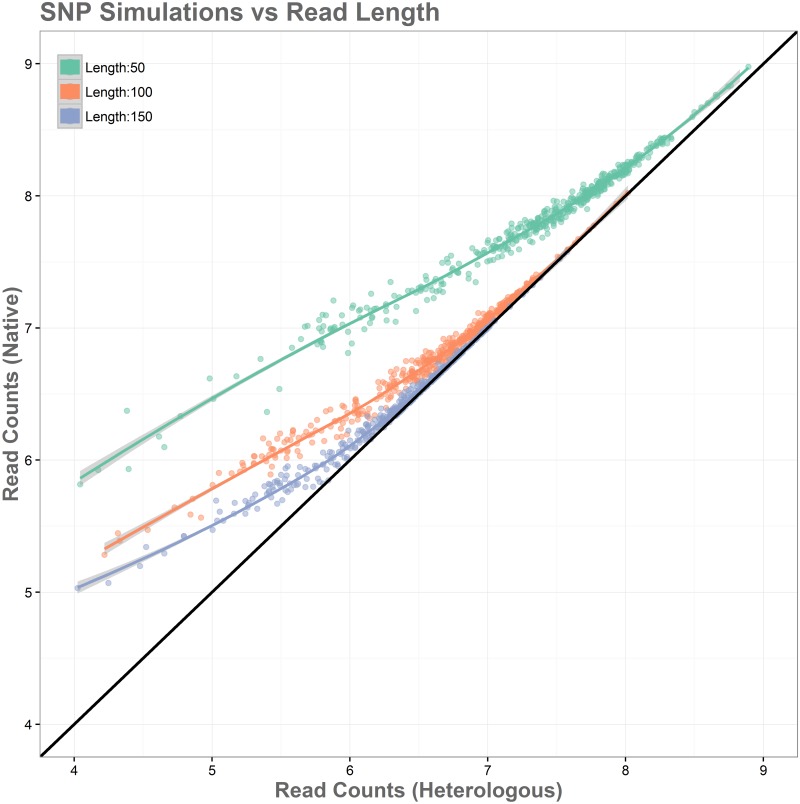
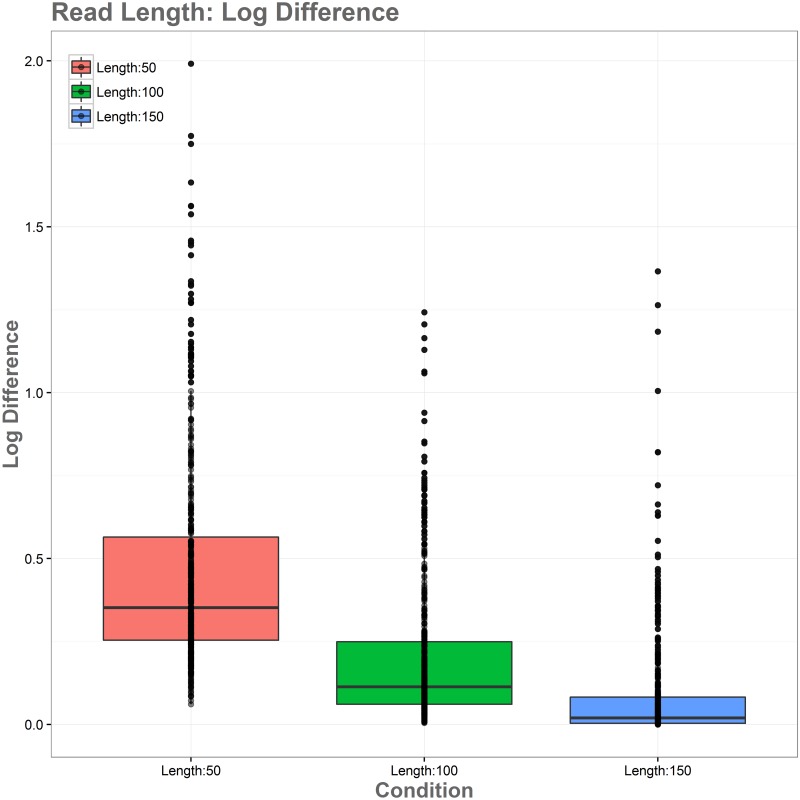
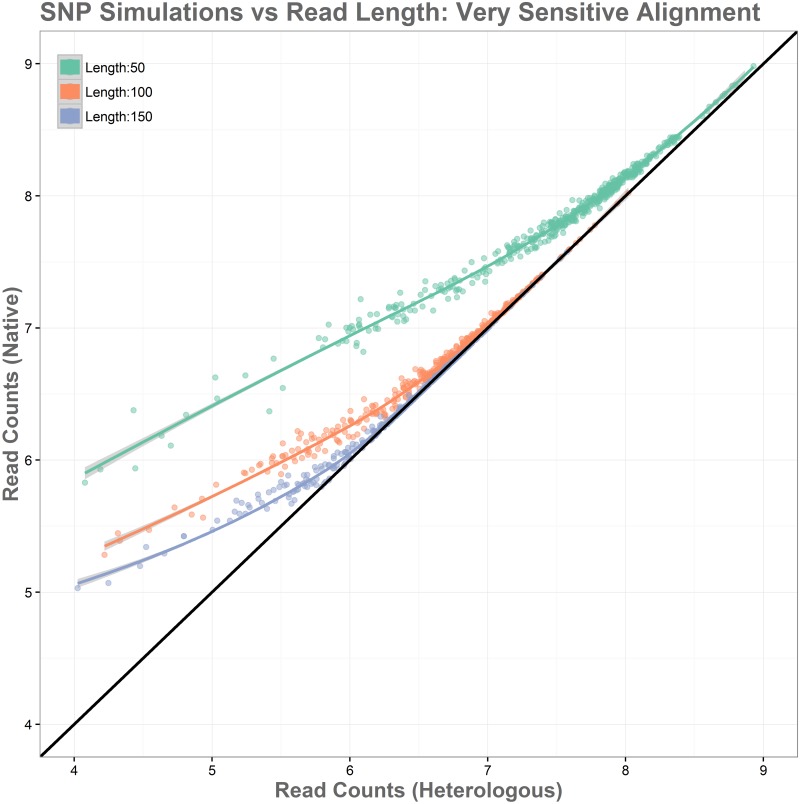
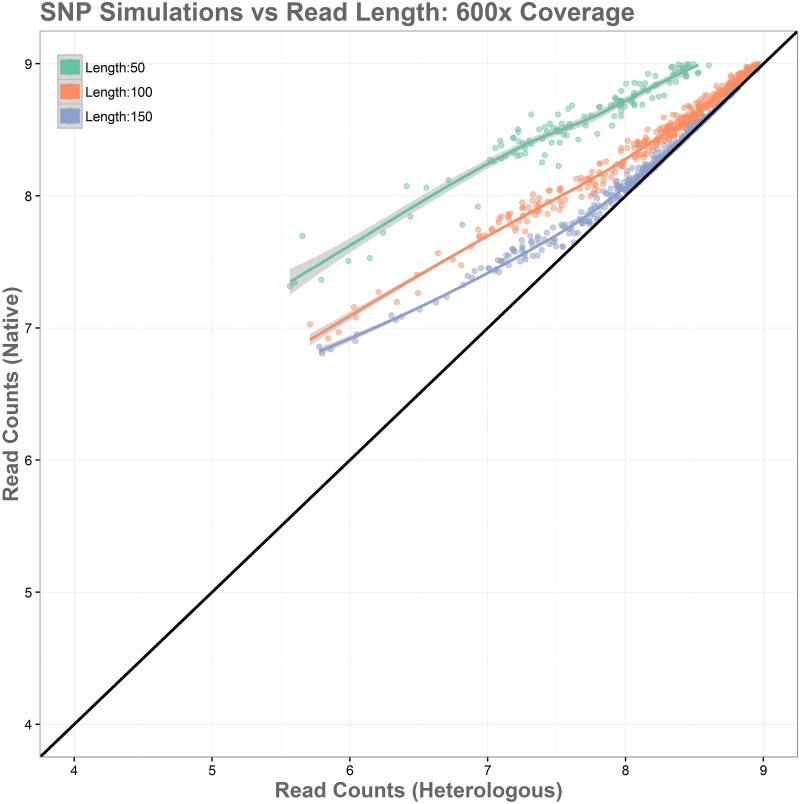
Sequence read alignment to a reference genome is a fundamental step in many genomics studies. Accuracy in this fundamental step is crucial for correct interpretation of biological data. In cases where two or more closely related bacterial strains are being studied, a common approach is to simply map reads from all strains to a common reference genome, whether because there is no closed reference for some strains or for ease of comparison. The assumption is that the differences between bacterial strains are insignificant enough that the results of differential expression analysis will not be influenced by choice of reference. Genes that are common among the strains under study are used for differential expression analysis, while the remaining genes, which may fail to express in one sample or the other because they are simply absent, are analyzed separately. In this study, we investigate the practice of using a common reference in transcriptomic analysis. We analyze two multi-strain transcriptomic data sets that were initially presented in the literature as comparisons based on a common reference, but which have available closed genomic sequence for all strains, allowing a detailed examination of the impact of reference choice. We provide a method for identifying regions that are most affected by non-native alignments, leading to false positives in differential expression analysis, and perform an in depth analysis identifying the extent of expression loss. We also simulate several data sets to identify best practices for non-native reference use.
将序列读数比对到参考基因组是许多基因组学研究中的基本步骤。这一基本步骤的准确性对于正确解读生物学数据至关重要。在研究两种或更多密切相关的细菌菌株的情况下,一种常见的方法是简单地将所有菌株的读数映射到一个共同的参考基因组,无论是因为某些菌株没有完整的参考基因组,还是为了便于比较。其假设是细菌菌株之间的差异足够小,以至于差异表达分析的结果不会受到参考选择的影响。在所研究的菌株中常见的基因用于差异表达分析,而其余的基因,可能由于根本不存在而在一个样本或另一个样本中未能表达,则单独进行分析。在本研究中,我们调查了在转录组分析中使用共同参考的做法。我们分析了两个多菌株转录组数据集,这些数据集最初在文献中是以基于共同参考的比较形式呈现的,但所有菌株都有可用的完整基因组序列,这使得我们能够详细检查参考选择的影响。我们提供了一种方法来识别受非天然比对影响最大的区域,这些区域会导致差异表达分析中的假阳性,并进行深入分析以确定表达损失的程度。我们还模拟了几个数据集,以确定使用非天然参考的最佳做法。