Persani Luca, Cangiano Biagio, Bonomi Marco
Division of Endocrine and Metabolic Diseases, Istituto Auxologico Italiano IRCCS, Milan, Italy.
Department of Clinical Sciences and Community Health, University of Milan, Milan, Italy.
Endocr Connect. 2019 Feb;8(2):R44-R54. doi: 10.1530/EC-18-0515.
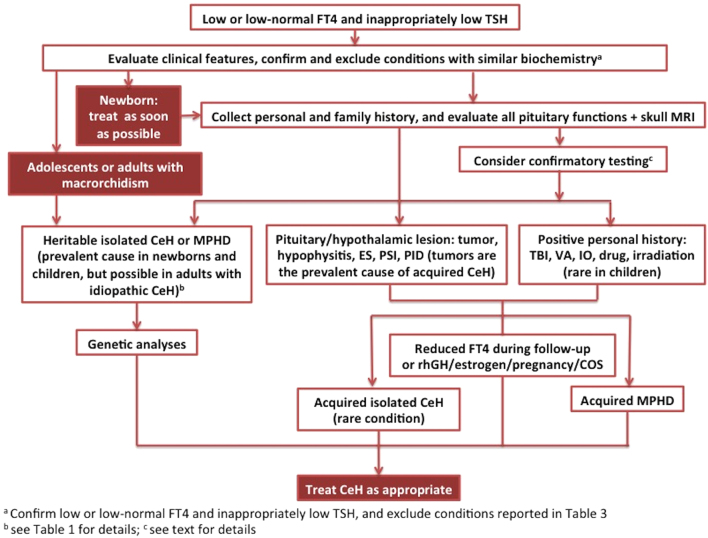
Central hypothyrodism (CeH) is a hypothyroid state caused by an insufficient stimulation by thyrotropin (TSH) of an otherwise normal thyroid gland. Several advancements, including the recent publication of expert guidelines for CeH diagnosis and management, have been made in recent years thus increasing the clinical awareness on this condition. Here, we reviewed the recent advancements and give expert opinions on critical issues. Indeed, CeH can be the consequence of various disorders affecting either the pituitary gland or the hypothalamus. Recent data enlarged the list of candidate genes for heritable CeH and a genetic origin may be the underlying cause for CeH discovered in pediatric or even adult patients without apparent pituitary lesions. This raises the doubt that the frequency of CeH may be underestimated. CeH is most frequently diagnosed as a consequence of the biochemical assessments in patients with hypothalamic/pituitary lesions. In contrast with primary hypothyroidism, low FT4 with low/normal TSH levels are the biochemical hallmark of CeH, and adequate thyroid hormone replacement leads to the suppression of residual TSH secretion. Thus, CeH often represents a clinical challenge because physicians cannot rely on the use of the 'reflex TSH strategy' for screening or therapy monitoring. Nevertheless, in contrast with general assumption, the finding of normal TSH levels may indicate thyroxine under-replacement in CeH patients. The clinical management of CeH is further complicated by the combination with multiple pituitary deficiencies, as the introduction of sex steroids or GH replacements may uncover latent forms of CeH or increase the thyroxine requirements.
中枢性甲状腺功能减退症(CeH)是一种甲状腺功能减退状态,由促甲状腺激素(TSH)对原本正常的甲状腺刺激不足所致。近年来取得了多项进展,包括最近发布了CeH诊断和管理的专家指南,从而提高了对这种疾病的临床认识。在此,我们回顾了最近的进展,并就关键问题给出专家意见。事实上,CeH可能是影响垂体或下丘脑的各种疾病的结果。最近的数据扩大了遗传性CeH候选基因的列表,并且遗传起源可能是在没有明显垂体病变的儿科甚至成年患者中发现的CeH的潜在原因。这引发了对CeH发病率可能被低估的怀疑。CeH最常因下丘脑/垂体病变患者的生化评估而被诊断。与原发性甲状腺功能减退症相反,低游离甲状腺素(FT4)伴低/正常促甲状腺激素(TSH)水平是CeH的生化标志,适当的甲状腺激素替代会导致残余TSH分泌受到抑制。因此,CeH常常代表一项临床挑战,因为医生不能依赖“反射性TSH策略”进行筛查或治疗监测。然而,与一般假设相反,TSH水平正常的发现可能表明CeH患者甲状腺素替代不足。CeH的临床管理因合并多种垂体功能减退而进一步复杂化,因为引入性类固醇或生长激素替代可能会发现潜在形式的CeH或增加甲状腺素需求。