Center for Bioinformatics, State Key Laboratory of Protein and Plant Gene Research, School of Life Sciences, Peking University, Beijing, 100871, People's Republic of China.
BMC Bioinformatics. 2019 Jan 15;20(1):28. doi: 10.1186/s12859-019-2597-8.
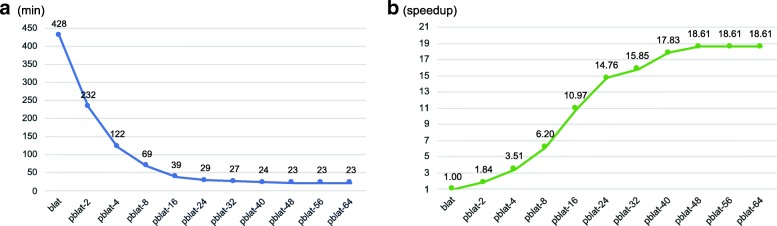
The blat is a widely used sequence alignment tool. It is especially useful for aligning long sequences and gapped mapping, which cannot be performed properly by other fast sequence mappers designed for short reads. However, the blat tool is single threaded and when used to map whole genome or whole transcriptome sequences to reference genomes this program can take days to finish, making it unsuitable for large scale sequencing projects and iterative analysis. Here, we present pblat (parallel blat), a parallelized blat algorithm with multithread and cluster computing support, which functions to rapidly fine map large scale DNA/RNA sequences against genomes.
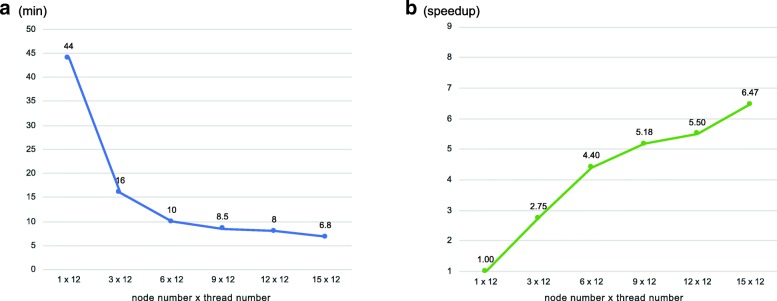
The pblat algorithm takes advantage of modern multicore processors and significantly reduces the run time with the number of threads used. pblat utilizes almost equal amount of memory as when running blat. The results generated by pblat are identical with those generated by blat. The pblat tool is easy to install and can run on Linux and Mac OS systems. In addition, we provide a cluster version of pblat (pblat-cluster) running on computing clusters with MPI support.
pblat is open source and free available for non-commercial users. It is easy to install and easy to use. pblat and pblat-cluster would facilitate the high-throughput mapping of large scale genomic and transcript sequences to reference genomes with both high speed and high precision.
blat 是一种广泛使用的序列比对工具。它特别适用于对齐长序列和缺口映射,而其他专为短读长设计的快速序列映射器无法正确执行这些操作。然而,blat 工具是单线程的,当用于将整个基因组或整个转录组序列映射到参考基因组时,该程序可能需要数天才能完成,因此不适合大规模测序项目和迭代分析。在这里,我们提出了 pblat(并行 blat),这是一种具有多线程和集群计算支持的并行 blat 算法,用于快速对大规模 DNA/RNA 序列进行精细映射到基因组上。
pblat 算法利用现代多核处理器,通过使用的线程数量显著缩短运行时间。pblat 利用的内存与 blat 运行时几乎相同。pblat 生成的结果与 blat 生成的结果完全一致。pblat 工具易于安装,可以在 Linux 和 Mac OS 系统上运行。此外,我们提供了一个带有 MPI 支持的计算集群上运行的 pblat 集群版本(pblat-cluster)。
pblat 是开源的,免费供非商业用户使用。它易于安装和使用。pblat 和 pblat-cluster 将有助于以高速和高精度将大规模基因组和转录序列映射到参考基因组。