Institute of Biotechnology of the Czech Academy of Sciences, BIOCEV, Průmyslová 595, 252 50 Vestec, Prague West, Czech Republic.
Int J Mol Sci. 2019 Jan 16;20(2):370. doi: 10.3390/ijms20020370.
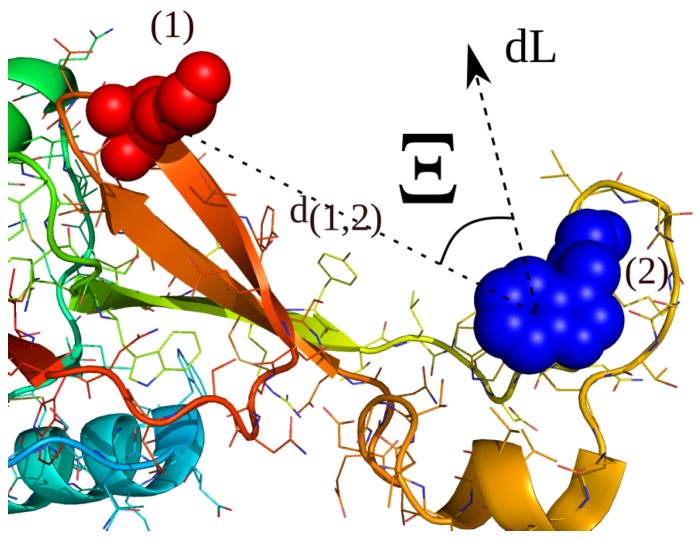
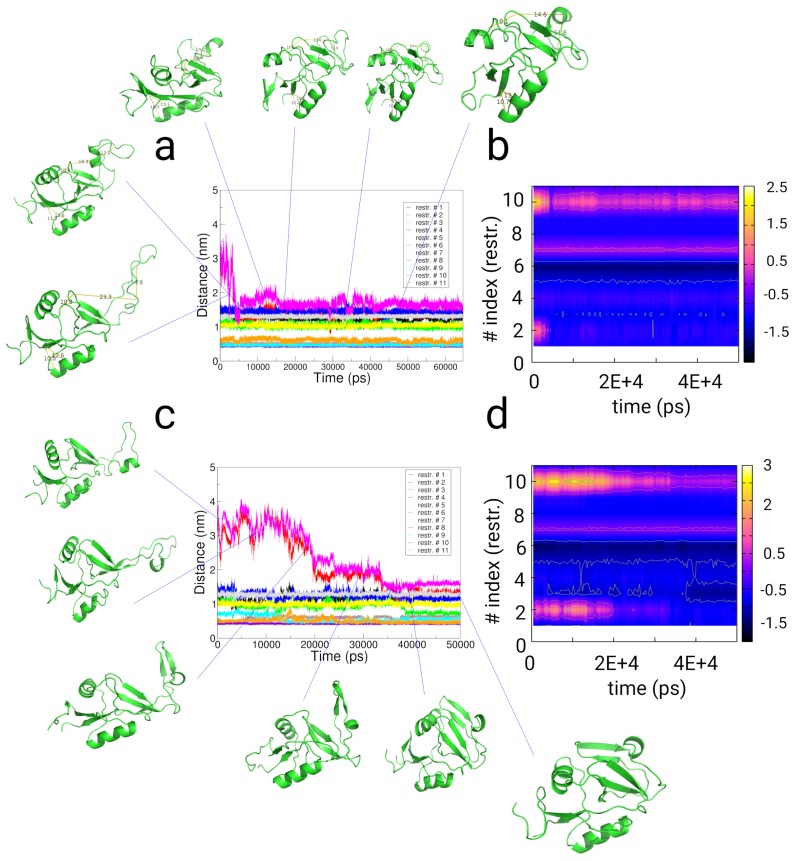
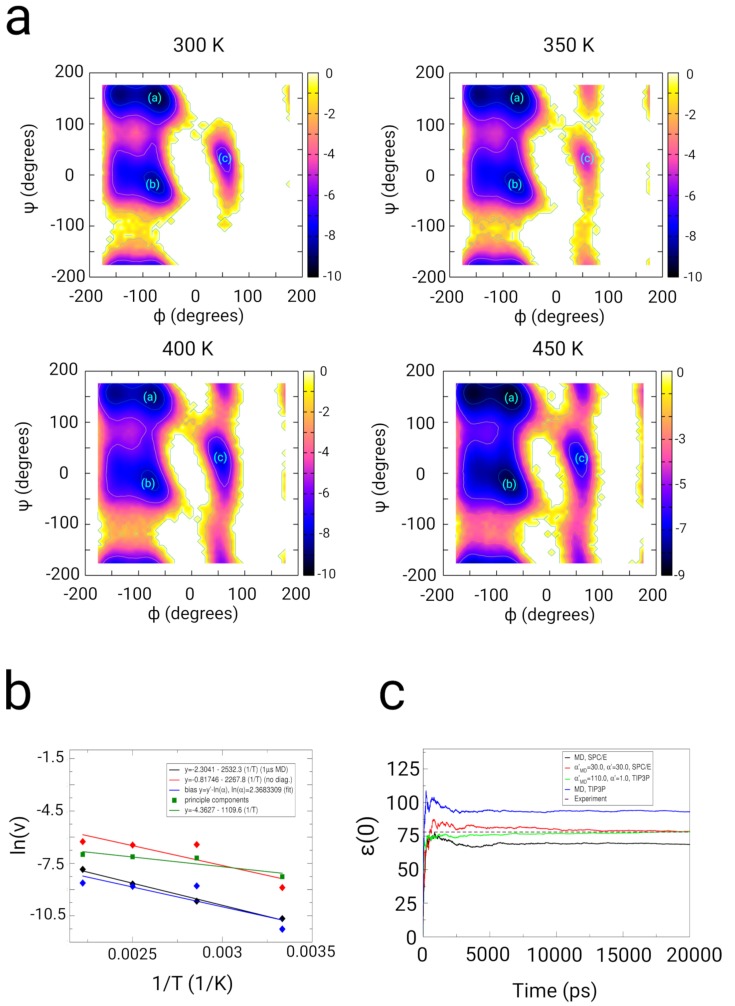
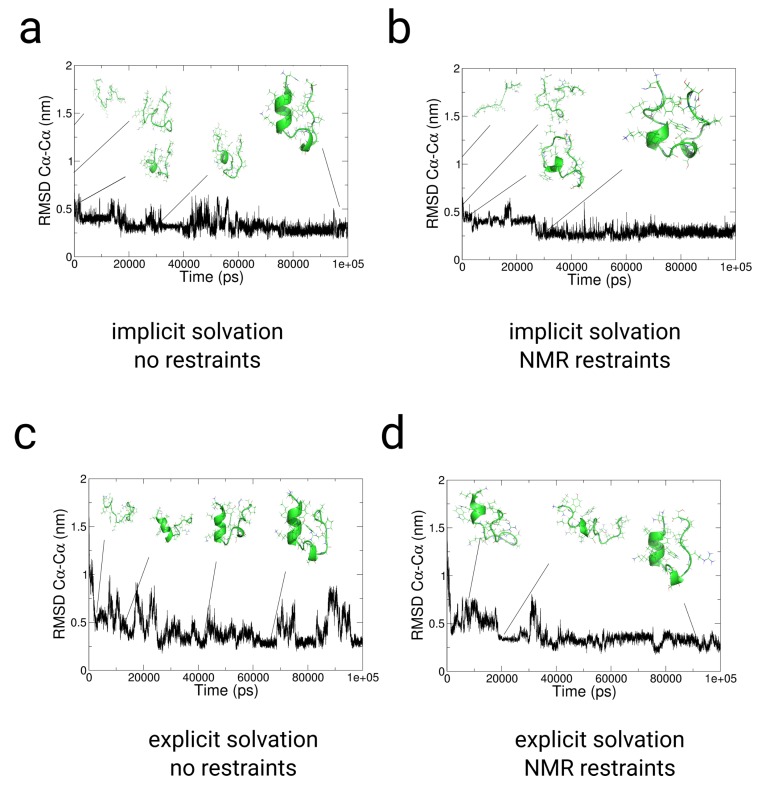
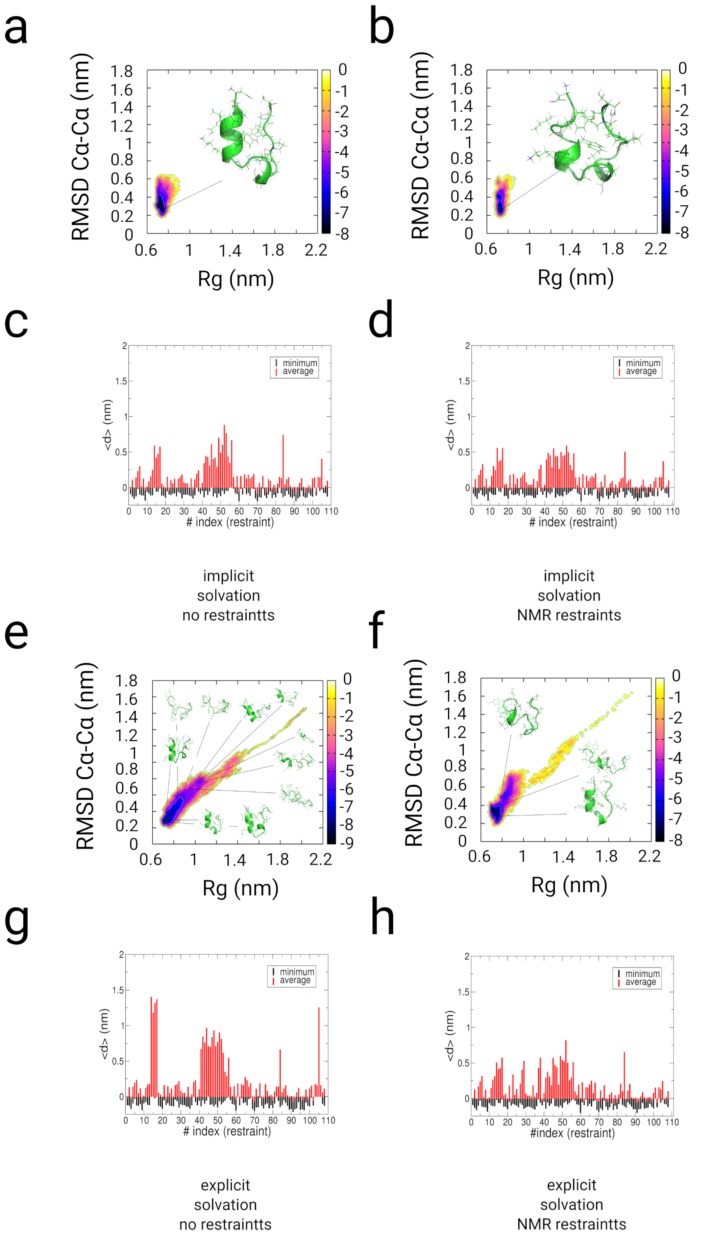
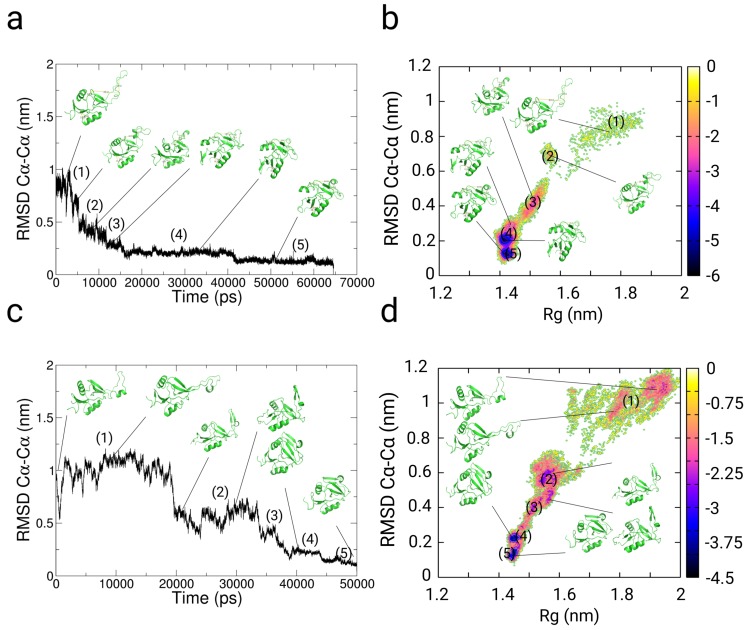
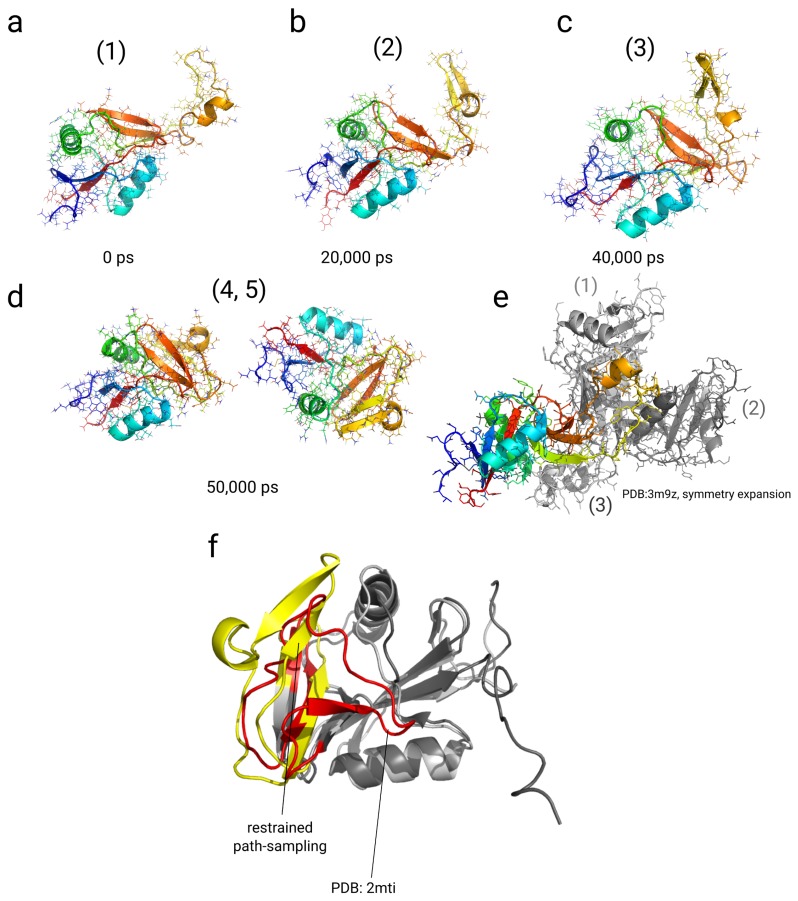
In this article, we present an enhanced sampling method based on a hybrid Hamiltonian which combines experimental distance restraints with a bias dependent from multiple path-dependent variables. This simulation method determines the bias-coordinates and does not require knowledge about reaction coordinates. The hybrid Hamiltonian accelerates the sampling of proteins, and, combined with experimental distance information, the technique considers the restraints adaptively and in dependency of the system's intrinsic dynamics. We validate the methodology on the dipole relaxation of two water models and the conformational landscape of dialanine. Using experimental NMR-restraint data, we explore the folding landscape of the TrpCage mini-protein and in a second example apply distance restraints from chemical crosslinking/mass spectrometry experiments for the sampling of the conformation space of the Killer Cell Lectin-like Receptor Subfamily B Member 1A (NKR-P1A). The new methodology has the potential to adaptively introduce experimental restraints without affecting the conformational space of the system along an ergodic trajectory. Since only a limited number of input- and no-order parameters are required for the setup of the simulation, the method is broadly applicable and has the potential to be combined with coarse-graining methods.
在本文中,我们提出了一种基于混合哈密顿量的增强采样方法,该方法将实验距离约束与依赖于多个路径相关变量的偏差相结合。这种模拟方法确定了偏差坐标,并且不需要关于反应坐标的知识。混合哈密顿量加速了蛋白质的采样,并且,结合实验距离信息,该技术自适应地考虑了约束,并且依赖于系统的固有动力学。我们在两个水分子模型的偶极弛豫和二丙氨酸的构象景观上验证了该方法的有效性。使用实验 NMR 约束数据,我们探索了 TrpCage 小蛋白的折叠景观,并在第二个示例中应用了来自化学交联/质谱实验的距离约束,以对 Killer Cell Lectin-like Receptor Subfamily B Member 1A (NKR-P1A)构象空间进行采样。新方法具有自适应引入实验约束的潜力,而不会沿着遍历轨迹影响系统的构象空间。由于设置模拟仅需要有限数量的输入和无序参数,因此该方法具有广泛的适用性,并且有可能与粗粒化方法相结合。