Department of Biology, New York University, New York, NY, 10003, USA.
Present address: BioQuant Center, Heidelberg University, Heidelberg, Germany.
BMC Genomics. 2019 Jan 17;20(1):54. doi: 10.1186/s12864-018-5368-4.
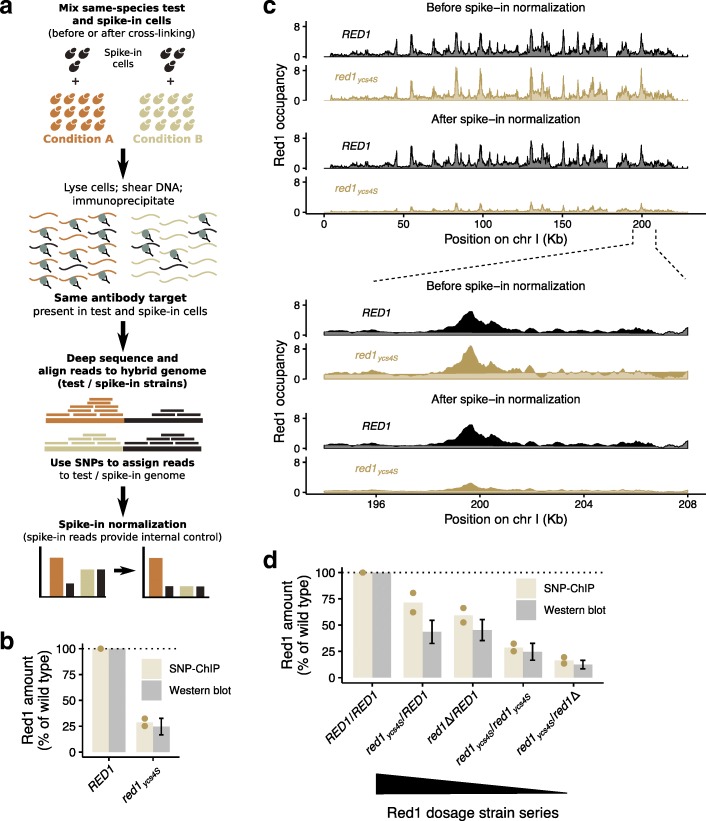
Chromatin-immunoprecipitation followed by sequencing (ChIP-seq) is the method of choice for mapping genome-wide binding of chromatin-associated factors. However, broadly applicable methods for between-sample comparisons are lacking.
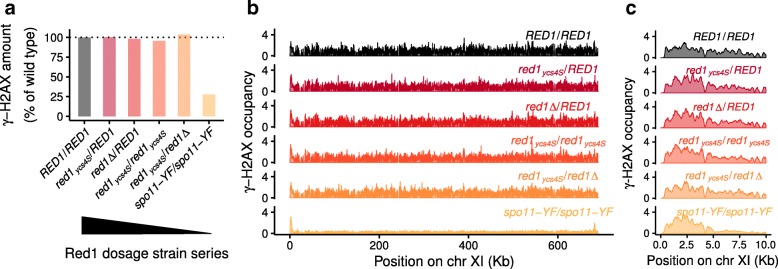
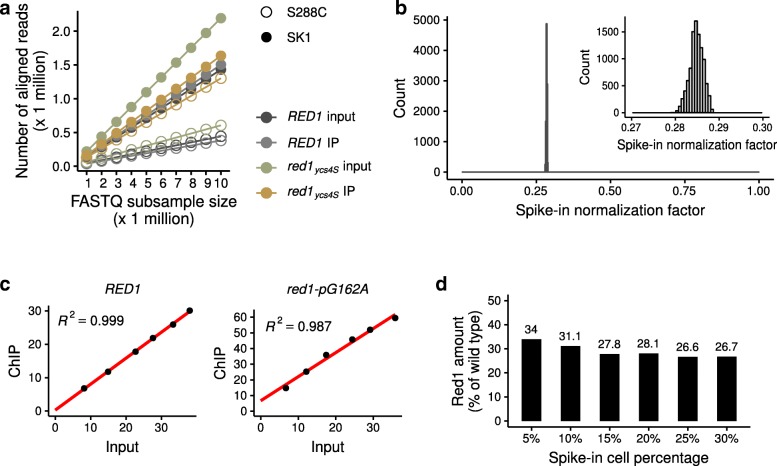
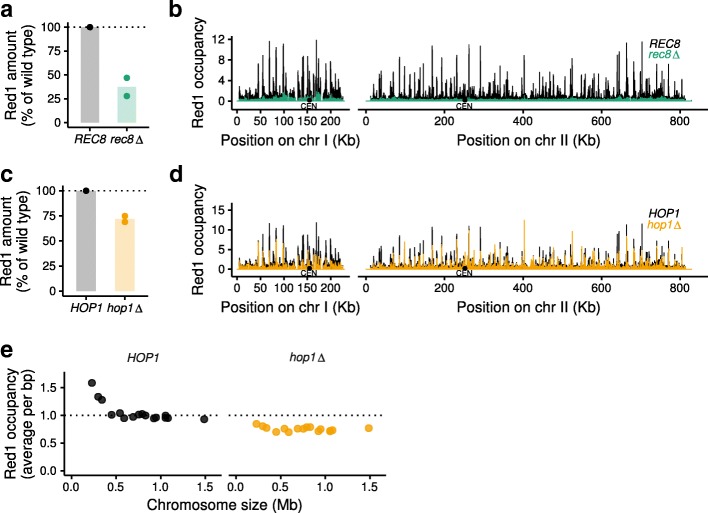
Here, we introduce SNP-ChIP, a method that leverages small-scale intra-species polymorphisms, mainly SNPs, for quantitative spike-in normalization of ChIP-seq results. Sourcing spike-in material from the same species ensures antibody cross-reactivity and physiological coherence, thereby eliminating two central limitations of traditional spike-in approaches. We show that SNP-ChIP is robust to changes in sequencing depth and spike-in proportions, and reliably identifies changes in overall protein levels, irrespective of changes in binding distribution. Application of SNP-ChIP to test cases from budding yeast meiosis allowed discovery of novel regulators of the chromosomal protein Red1 and quantitative analysis of the DNA-damage associated histone modification γ-H2AX.
SNP-ChIP is fully compatible with the intra-species diversity of humans and most model organisms and thus offers a general method for normalizing ChIP-seq results.
染色质免疫沉淀测序(ChIP-seq)是用于绘制全基因组染色质相关因子结合图谱的首选方法。然而,缺乏适用于样本间比较的广泛方法。
在这里,我们介绍了 SNP-ChIP 方法,该方法利用种内小规模的多态性,主要是 SNP,对 ChIP-seq 结果进行定量 Spike-in 标准化。从同一物种中获取 Spike-in 材料可确保抗体的交叉反应性和生理一致性,从而消除了传统 Spike-in 方法的两个核心限制。我们表明,SNP-ChIP 对测序深度和 Spike-in 比例的变化具有鲁棒性,并且可以可靠地识别整体蛋白质水平的变化,而与结合分布的变化无关。将 SNP-ChIP 应用于芽殖酵母减数分裂的测试案例,发现了 Red1 染色体蛋白的新调控因子,并对与 DNA 损伤相关的组蛋白修饰 γ-H2AX 进行了定量分析。
SNP-ChIP 完全兼容人类和大多数模式生物的种内多样性,因此为 ChIP-seq 结果的标准化提供了一种通用方法。