Zahrt Andrew F, Henle Jeremy J, Rose Brennan T, Wang Yang, Darrow William T, Denmark Scott E
Roger Adams Laboratory, Department of Chemistry, University of Illinois, Urbana, IL 61801, USA.
Science. 2019 Jan 18;363(6424). doi: 10.1126/science.aau5631.
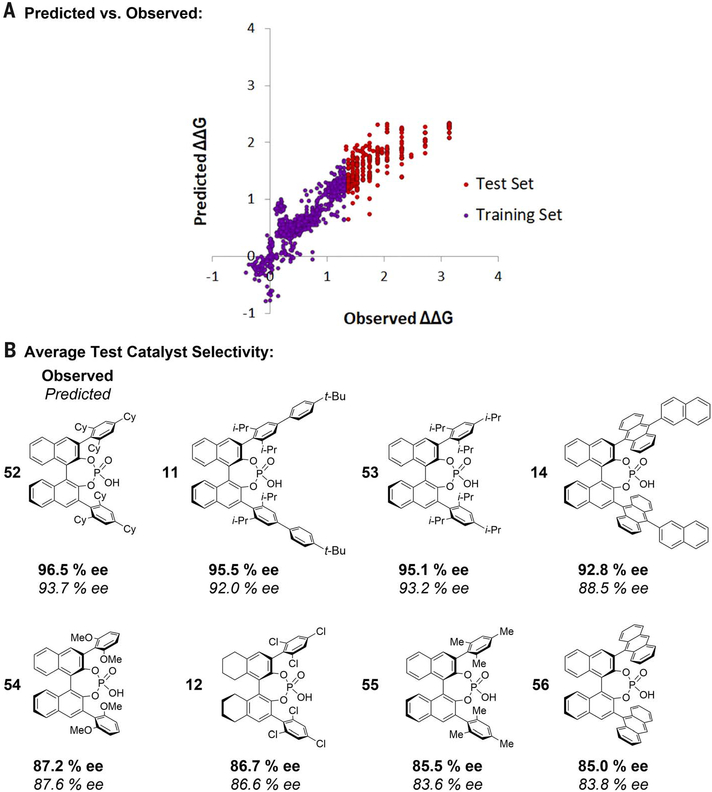
Catalyst design in asymmetric reaction development has traditionally been driven by empiricism, wherein experimentalists attempt to qualitatively recognize structural patterns to improve selectivity. Machine learning algorithms and chemoinformatics can potentially accelerate this process by recognizing otherwise inscrutable patterns in large datasets. Herein we report a computationally guided workflow for chiral catalyst selection using chemoinformatics at every stage of development. Robust molecular descriptors that are agnostic to the catalyst scaffold allow for selection of a universal training set on the basis of steric and electronic properties. This set can be used to train machine learning methods to make highly accurate predictive models over a broad range of selectivity space. Using support vector machines and deep feed-forward neural networks, we demonstrate accurate predictive modeling in the chiral phosphoric acid-catalyzed thiol addition to -acylimines.
在不对称反应开发中,传统上催化剂设计是由经验主义驱动的,实验人员试图定性地识别结构模式以提高选择性。机器学习算法和化学信息学可以通过识别大型数据集中难以理解的模式来潜在地加速这一过程。在此,我们报告了一种在开发的每个阶段使用化学信息学进行手性催化剂选择的计算指导工作流程。对催化剂支架不敏感的稳健分子描述符允许基于空间和电子性质选择通用训练集。该训练集可用于训练机器学习方法,以在广泛的选择性空间内建立高精度预测模型。使用支持向量机和深度前馈神经网络,我们在手性磷酸催化硫醇加成到酰亚胺的反应中展示了准确的预测建模。