Department of Swine Infectious Diseases, Shanghai Veterinary Research Institute, Chinese Academy of Agricultural Sciences, Shanghai, China.
Shanghai Key laboratory of Veterinary Biotechnology, School of Agriculture and Biology, Shanghai JiaoTong University, Shanghai, China.
Infect Genet Evol. 2019 Apr;69:153-165. doi: 10.1016/j.meegid.2019.01.022. Epub 2019 Jan 21.
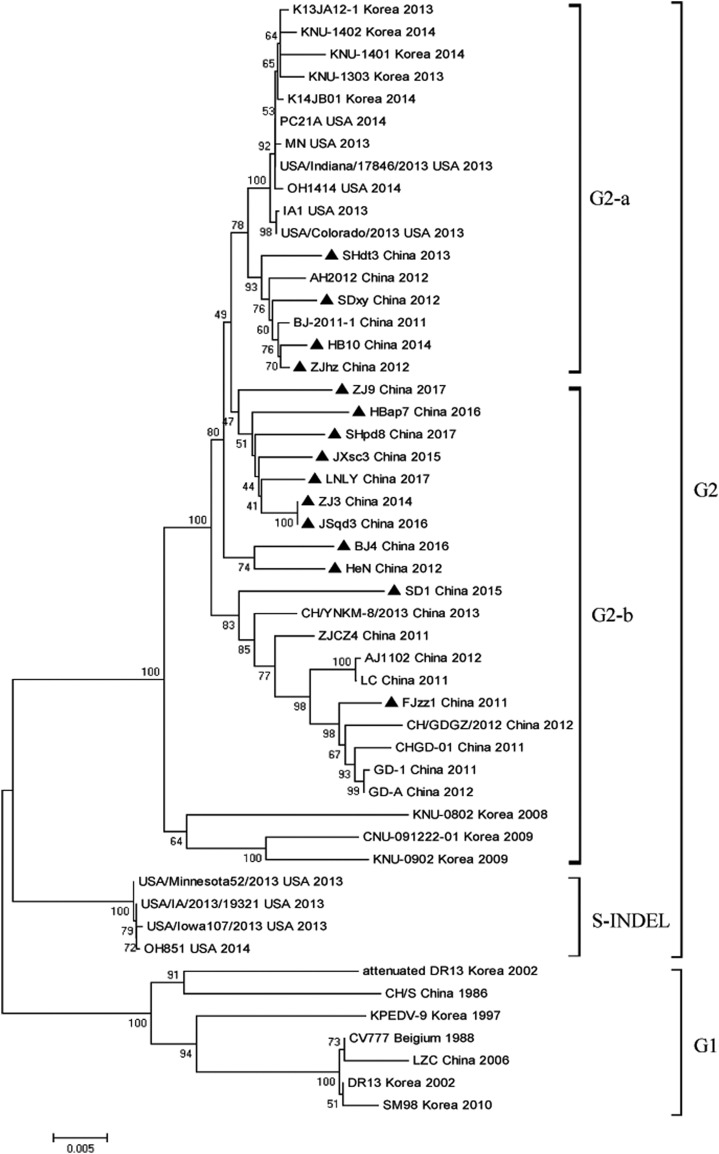
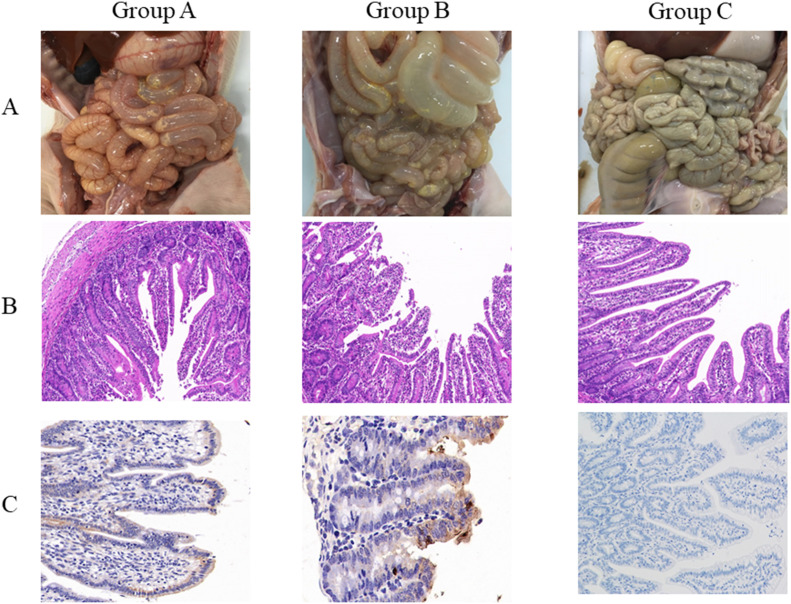
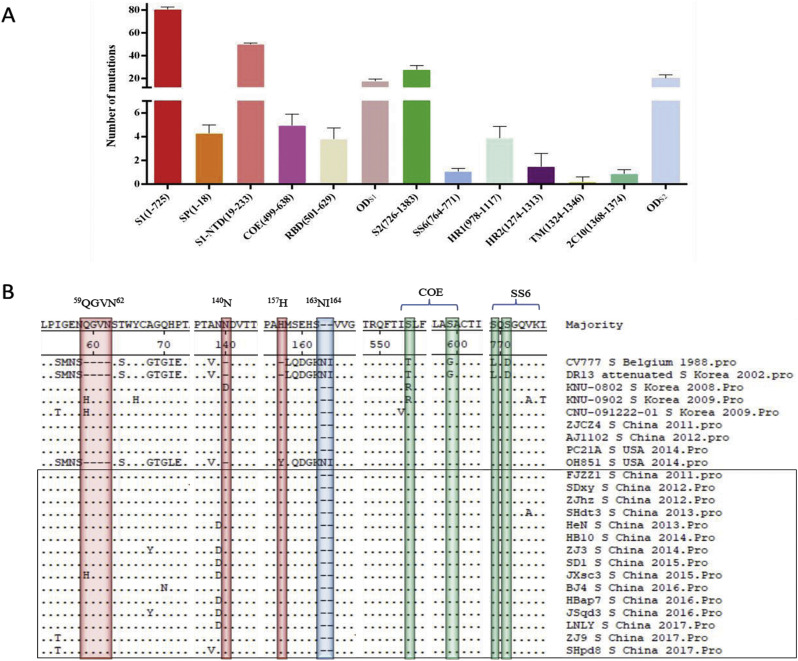
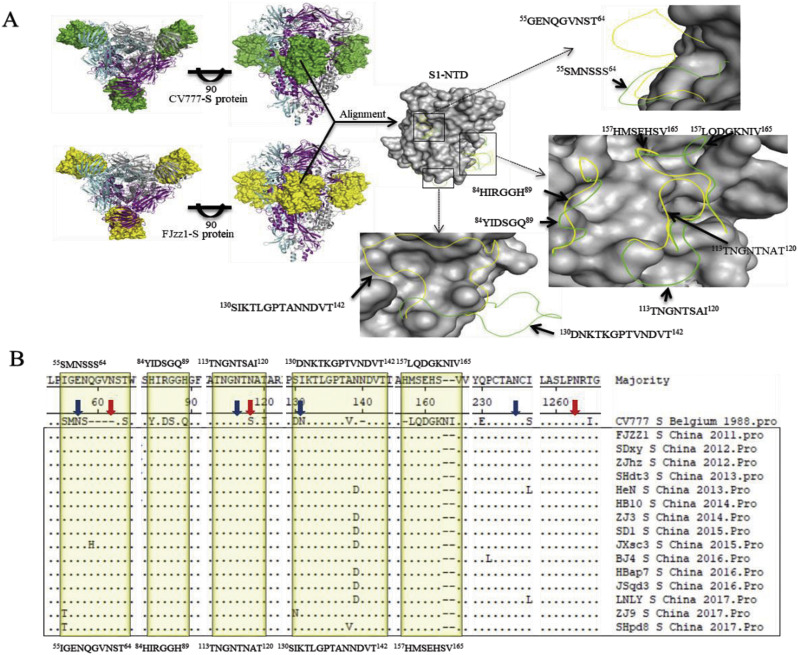
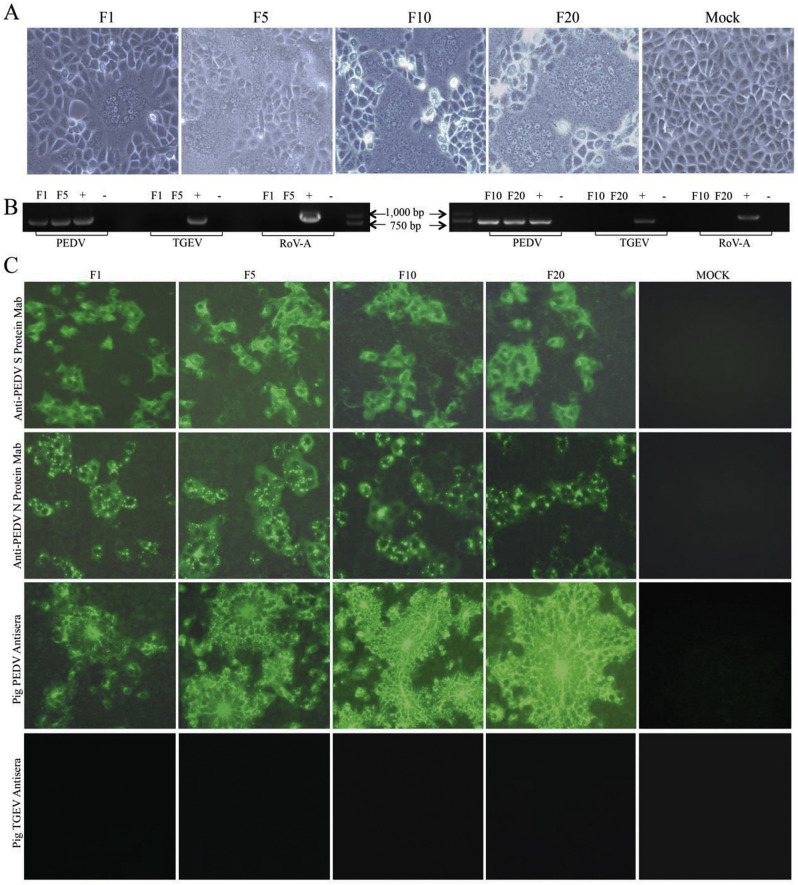
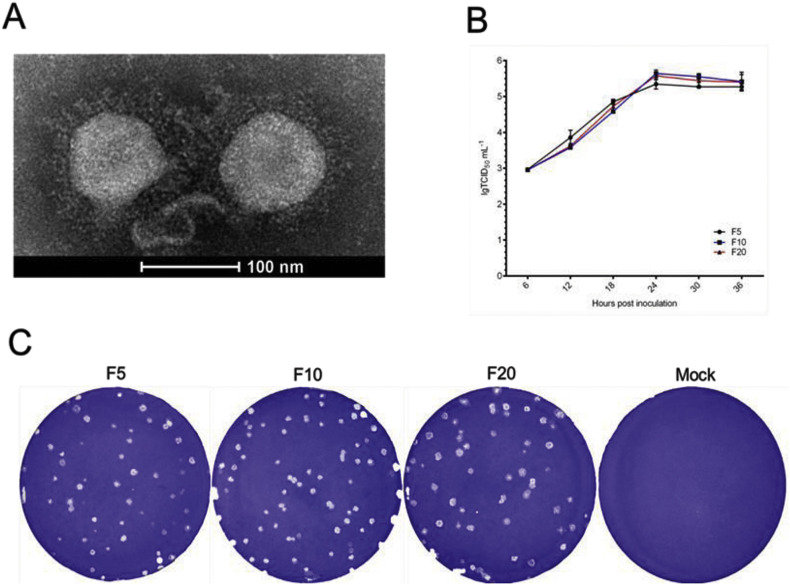
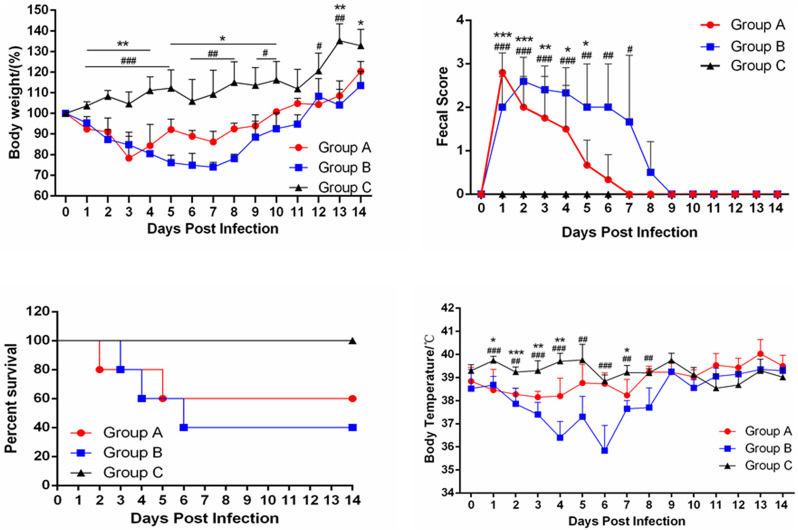
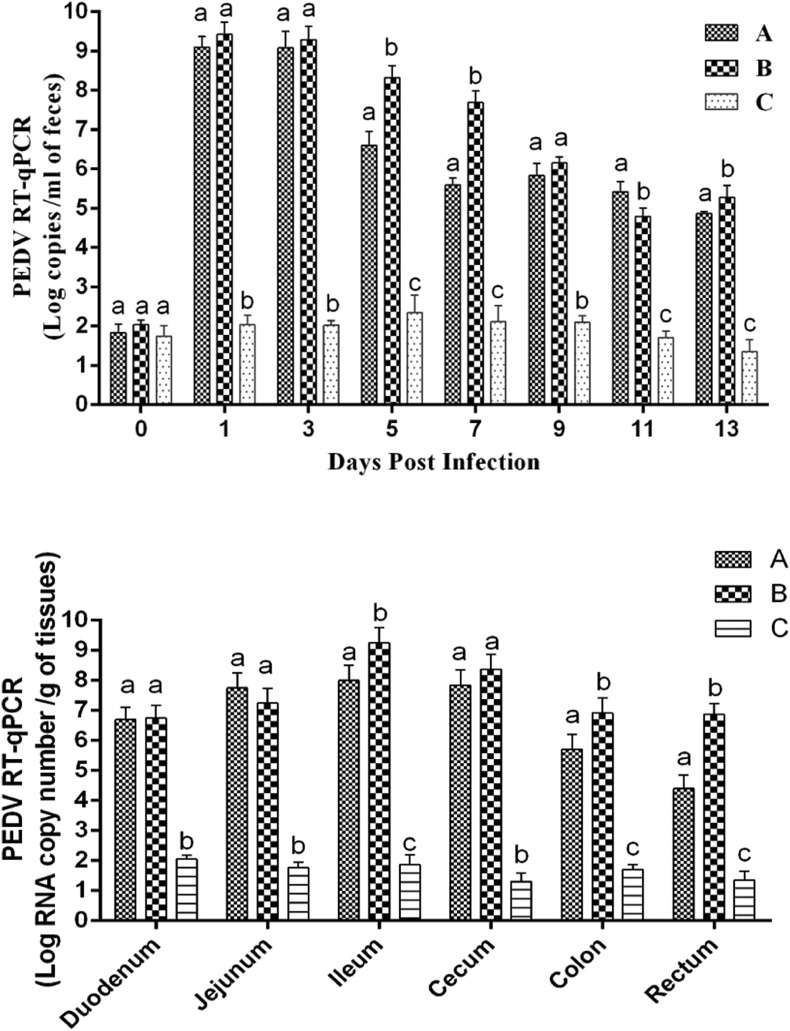
In recent years, the outbreaks of porcine epidemic diarrhea (PED) caused by the highly virulent porcine epidemic diarrhea virus (PEDV) variants occurred frequently in China, resulting in severe economic impacts to the pork industry. In this study, we selected and analyzed the genetic evolution of 15 PEDV representative strains that were identified in fecal samples of diarrheic piglets in 10 provinces and cities during 2011-2017. The phylogenetic analysis indicated that all the 15 PEDV isolates clustered into G2 genotype associated with the current circulating strains. Compared with the genome of the prototype strain CV777, these strains had 103-120 amino acid mutations in their S proteins, most of which were in the N terminal domain of S1 (S1-NTD). We also found 37 common mutations in all these 15 strains, although these strains shared 96.9-99.7% nucleotide homology and 96.3-99.8% amino acid homology in the S protein compared with the other original pandemic strains. Computational analysis showed that these mutations may lead to remarkable changes in the conformational structure and asparagine (N)-linked glycosylation sites of S1-NTD, which may be associated with the altered pathogenicity of these variant PEDV strains. We evaluated the pathogenicity of the PEDV strain FJzz1 in piglets through oral and intramuscular infection routes. Compared with oral infection, intramuscular infection could also cause typical clinical signs but with a slightly delayed onset, confirming that the variant PEDV isolate FJzz1 was highly pathogenic to suckling piglets. In conclusion, we analyzed the genetic variation and pathogenicity of the emerging PEDV isolates of China, indicating that G2 variant PEDV strains as the main prevalent strains that may mutate continually. This study shows the necessity of monitoring the molecular epidemiology and the etiological characteristics of the epidemic PEDV isolates, which may help better control the PED outbreaks.
近年来,中国频繁爆发由高致病性猪流行性腹泻病毒(PEDV)变异株引起的猪流行性腹泻(PED),给养猪业造成了严重的经济影响。在本研究中,我们选择并分析了 2011 年至 2017 年间从中国 10 个省、市腹泻仔猪粪便中鉴定出的 15 株 PEDV 代表株的遗传进化。系统进化分析表明,所有 15 株 PEDV 分离株均聚类到与当前流行株相关的 G2 基因型。与原型株 CV777 的基因组相比,这些分离株的 S 蛋白有 103-120 个氨基酸突变,其中大多数位于 S1 蛋白的 N 端结构域(S1-NTD)。我们还发现这 15 株分离株共有 37 个共同突变,尽管与其他原始大流行株相比,这些分离株的 S 蛋白核苷酸同源性为 96.9-99.7%,氨基酸同源性为 96.3-99.8%。计算分析表明,这些突变可能导致 S1-NTD 构象结构和天冬酰胺(N)连接糖基化位点发生显著变化,这可能与这些变异 PEDV 分离株致病性改变有关。我们通过口服和肌肉注射感染途径评估了 PEDV 株 FJzz1 在仔猪中的致病性。与口服感染相比,肌肉感染也可引起典型的临床症状,但发病时间稍晚,证实变异 PEDV 分离株 FJzz1 对仔猪具有高度致病性。综上所述,我们分析了中国新兴 PEDV 分离株的遗传变异和致病性,表明 G2 变异 PEDV 株是主要流行株,可能会持续发生突变。本研究表明有必要监测流行 PEDV 分离株的分子流行病学和病因学特征,这有助于更好地控制 PED 暴发。