Department of Cardiovascular and Internal Medicine, Kanazawa University Graduate School of Medicine, Kanazawa, Japan.
Department of Medical Biochemistry, Osaka University Graduate School of Frontier Biosciences, Suita, Japan.
ESC Heart Fail. 2019 Apr;6(2):406-415. doi: 10.1002/ehf2.12410. Epub 2019 Jan 28.
Cardiac myosin light chain kinase (cMLCK) phosphorylates ventricular myosin regulatory light chain 2 (MLC2v) and regulates sarcomere and cardiomyocyte organization. However, few data exist regarding the relationship between cMLCK mutations and MLC2v phosphorylation, particularly in terms of developing familial dilated cardiomyopathy (DCM) in whom cMLCK gene mutations were identified. The purpose of the present study was to investigate functional consequences of cMLCK mutations in DCM patients.
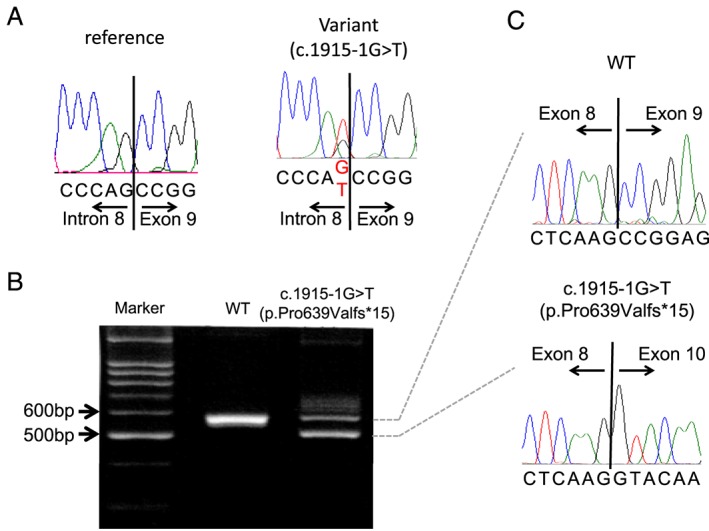
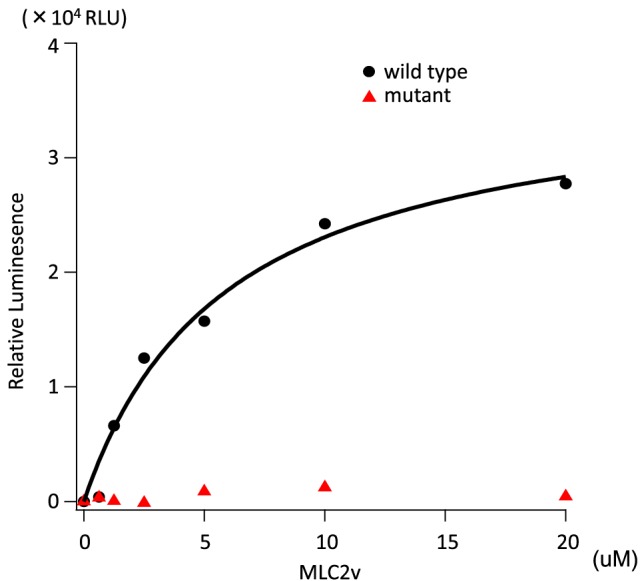
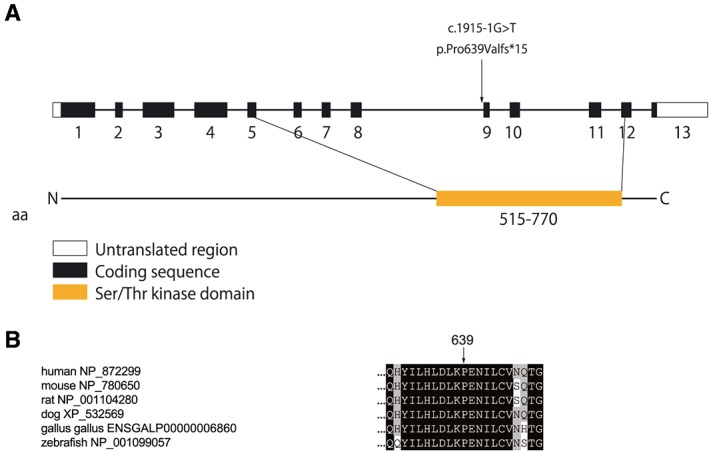
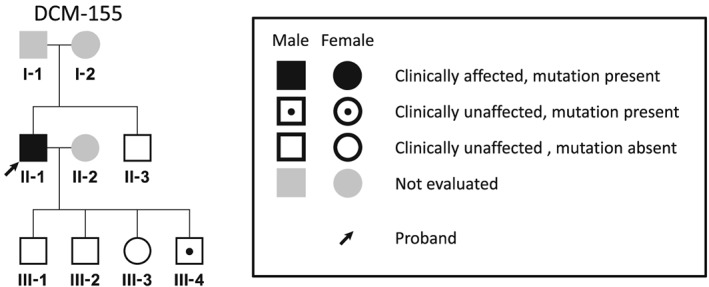
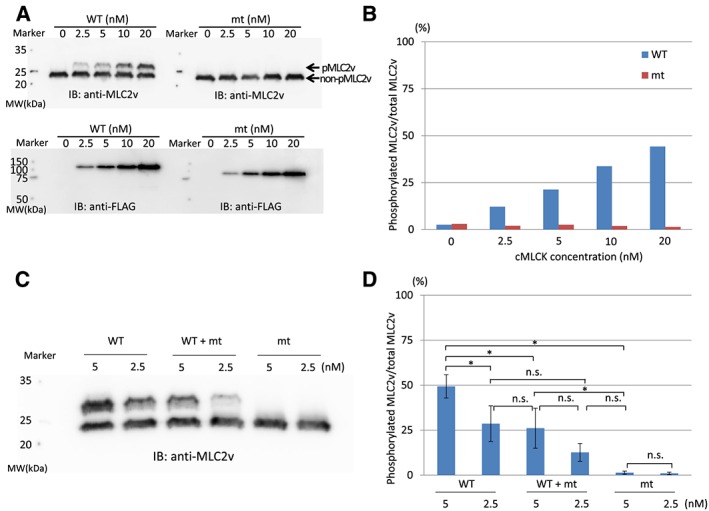
The diagnosis of DCM was based on the patients' history and on echocardiography. We screened cMLCK gene mutations in DCM probands with high resolution melting analysis. Known DCM-causing genes mutations were excluded by exome sequencing of family members. MLC2v phosphorylation was analysed by Phos-tag sodium dodecyl sulfate-polyacrylamide gel electrophoresis assays. We also performed ADP-Glo assays for determining the total amount of adenosine triphosphate used in the kinase reaction. Unrelated DCM probands (109 males and 40 females) were enrolled in this study, of which 16 were familial and 133 sporadic. By mutation screening, a truncation variant of c1915-1 g>t (p.Pro639Valfs15) was identified, which was not detected in 400 chromosomes of 200 healthy volunteers; it is listed in the Human Genetic Variation Database with an allele frequency < 0.001. In the proband, the presence of mutations in known DCM-causing genes was excluded with exome analysis. Familial analysis identified a 19-year-old male carrier who manifested slight left ventricular dilation with preserved systolic function. Phosphorylation assays analysed by Phos-tag SDS-PAGE revealed that the identified p.Pro639Valfs15 mutation results in a complete lack of kinase activity, although it did not affect wild-type cMLCK activity. ADP-Glo assays confirmed that the mutant cMLCK had no kinase activity, whereas wild-type cMLCK had a Km value of 5.93 ± 1.47 μM and a V of 1.28 ± 0.03 mol/min/mol kinase.
These results demonstrate that a truncation mutation in the cMLCK gene p.Pro639Valfs*15 can be associated with significant impairment of MLC2v phosphorylation and possibly with development of DCM, although a larger study of DCM patients is required to determine the prevalence of this mutation and further strengthen its association with disease development.
心肌球蛋白轻链激酶(cMLCK)磷酸化心室肌球蛋白调节轻链 2(MLC2v),并调节肌节和心肌细胞的组织。然而,关于 cMLCK 突变与 MLC2v 磷酸化之间的关系,特别是在发现 cMLCK 基因突变的家族性扩张型心肌病(DCM)患者中,相关数据很少。本研究的目的是研究 DCM 患者中 cMLCK 突变的功能后果。
DCM 的诊断基于患者的病史和超声心动图。我们通过高分辨率熔解分析筛选 DCM 先证者的 cMLCK 基因突变。通过对家族成员进行外显子组测序排除已知的 DCM 致病基因突变。通过 Phos-tag 十二烷基硫酸钠-聚丙烯酰胺凝胶电泳(SDS-PAGE)分析 MLC2v 磷酸化。我们还进行了 ADP-Glo 测定以确定激酶反应中使用的三磷酸腺苷总量。本研究纳入了 16 名家族性和 133 名散发性无关 DCM 先证者(109 名男性和 40 名女性)。通过突变筛选,发现了 c1915-1 g>t(p.Pro639Valfs15)的截断变异,该变异在 200 名健康志愿者的 400 条染色体中均未检出;它被列入人类遗传变异数据库,等位基因频率<0.001。在先证者中,外显子组分析排除了已知的 DCM 致病基因突变。家族分析发现一名 19 岁男性携带者,表现为轻度左心室扩张伴收缩功能正常。通过 Phos-tag SDS-PAGE 分析的磷酸化分析表明,鉴定的 p.Pro639Valfs15 突变导致激酶活性完全丧失,尽管它不影响野生型 cMLCK 活性。ADP-Glo 测定证实突变型 cMLCK 没有激酶活性,而野生型 cMLCK 的 Km 值为 5.93±1.47 μM,V 值为 1.28±0.03 mol/min/mol 激酶。
这些结果表明,cMLCK 基因 p.Pro639Valfs*15 的截断突变可导致 MLC2v 磷酸化显著受损,并可能导致 DCM 发生,尽管需要对 DCM 患者进行更大规模的研究以确定该突变的患病率并进一步加强其与疾病发展的相关性。