Institute for Experimental Medical Research, Oslo University Hospital and University of Oslo, Oslo, Norway.
KG Jebsen Center for Cardiac Research, University of Oslo, Oslo, Norway.
J Physiol. 2019 Apr;597(7):1833-1853. doi: 10.1113/JP277273. Epub 2019 Feb 27.
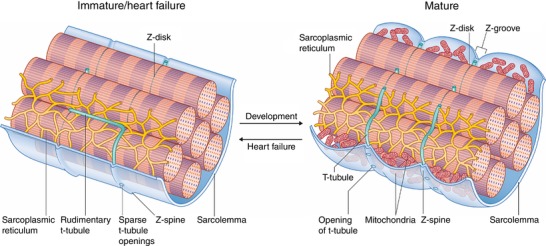
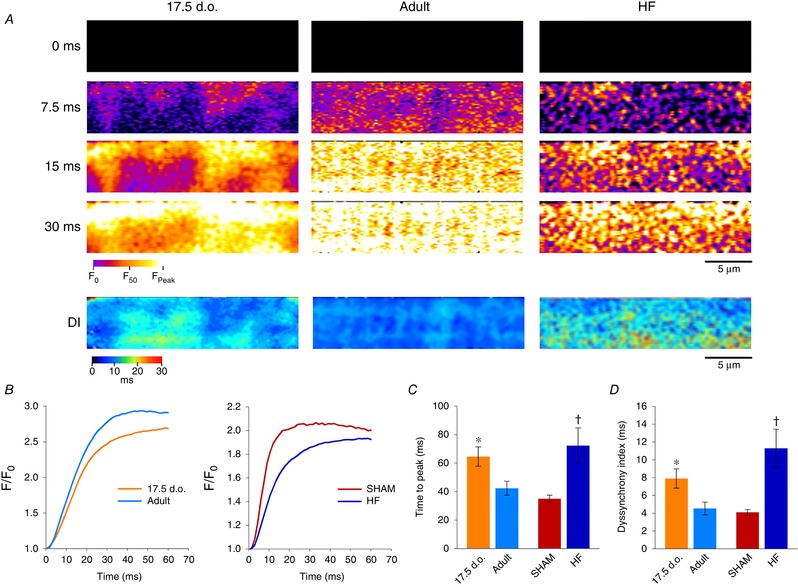
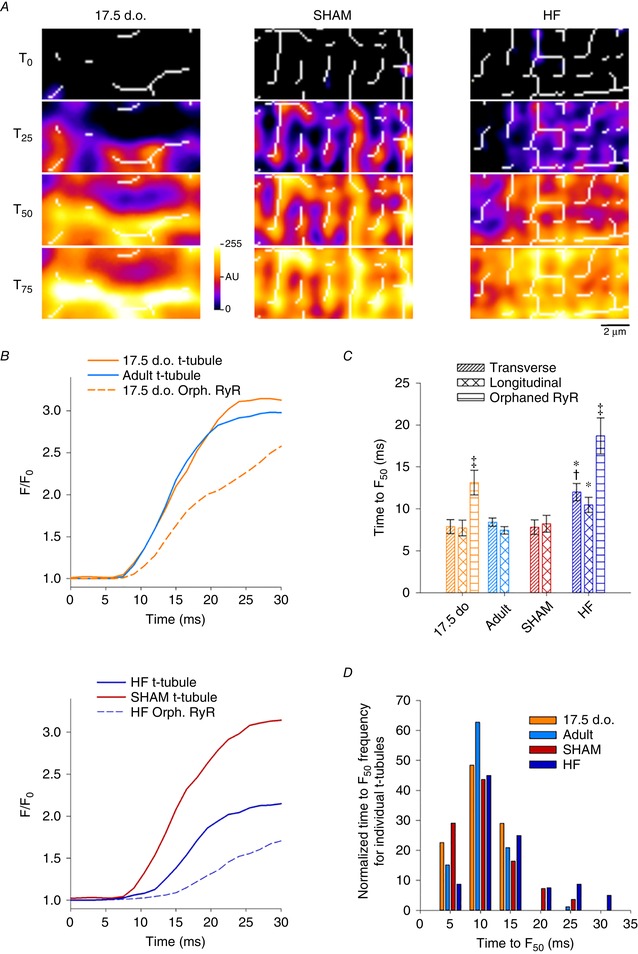
As reactivation of the fetal gene program has been implicated in pathological remodelling during heart failure (HF), we examined whether cardiomyocyte subcellular structure and function revert to an immature phenotype during this disease. Surface and internal membrane structures appeared gradually during development, and returned to a juvenile state during HF. Similarly, dyadic junctions between the cell membrane and sarcoplasmic reticulum were progressively 'packed' with L-type Ca channels and ryanodine receptors during development, and 'unpacked' during HF. Despite similarities in subcellular structure, dyads were observed to be functional from early developmental stages, but exhibited an impaired ability to release Ca in failing cardiomyocytes. Thus, while immature and failing cardiomyocytes share similarities in subcellular structure, these do not fully account for the marked impairment of Ca homeostasis observed in HF.
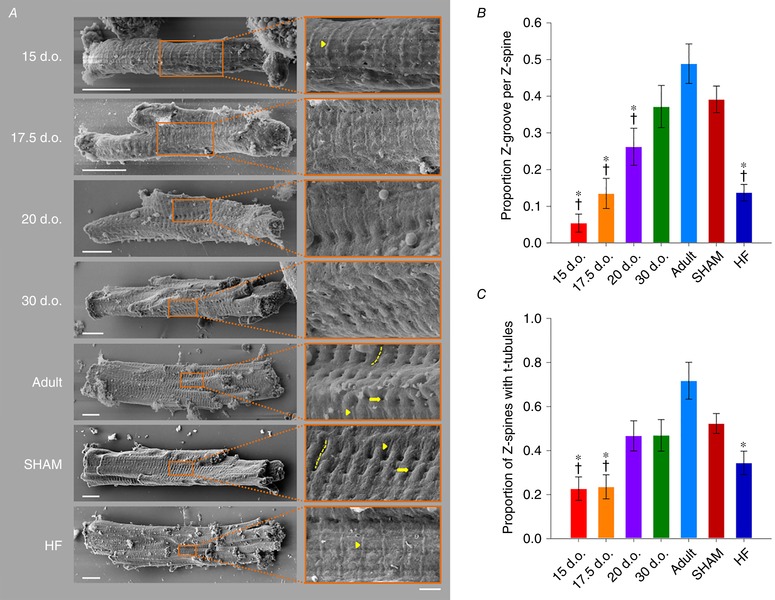
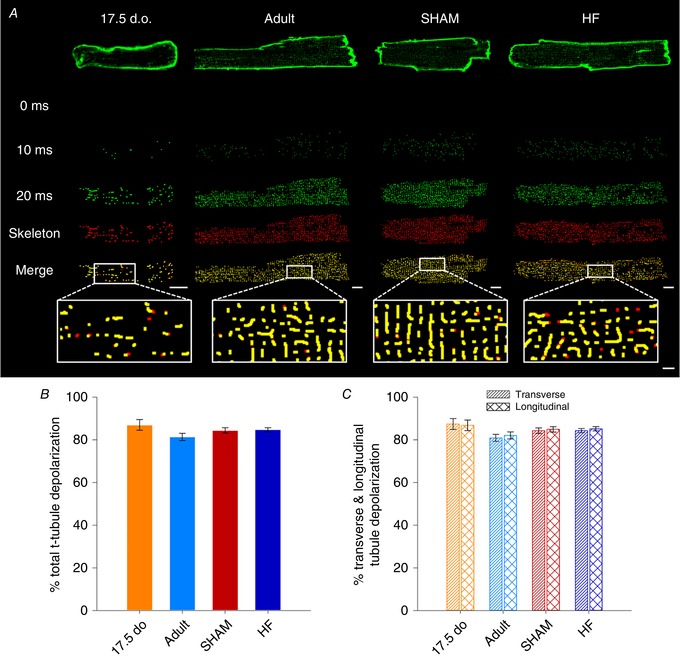
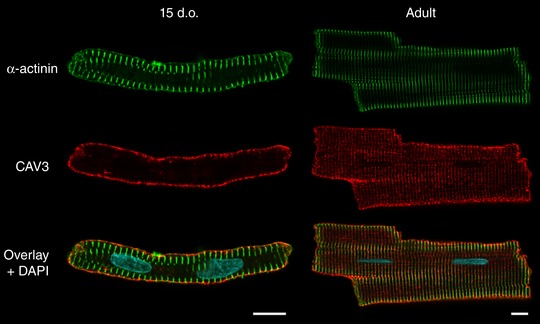
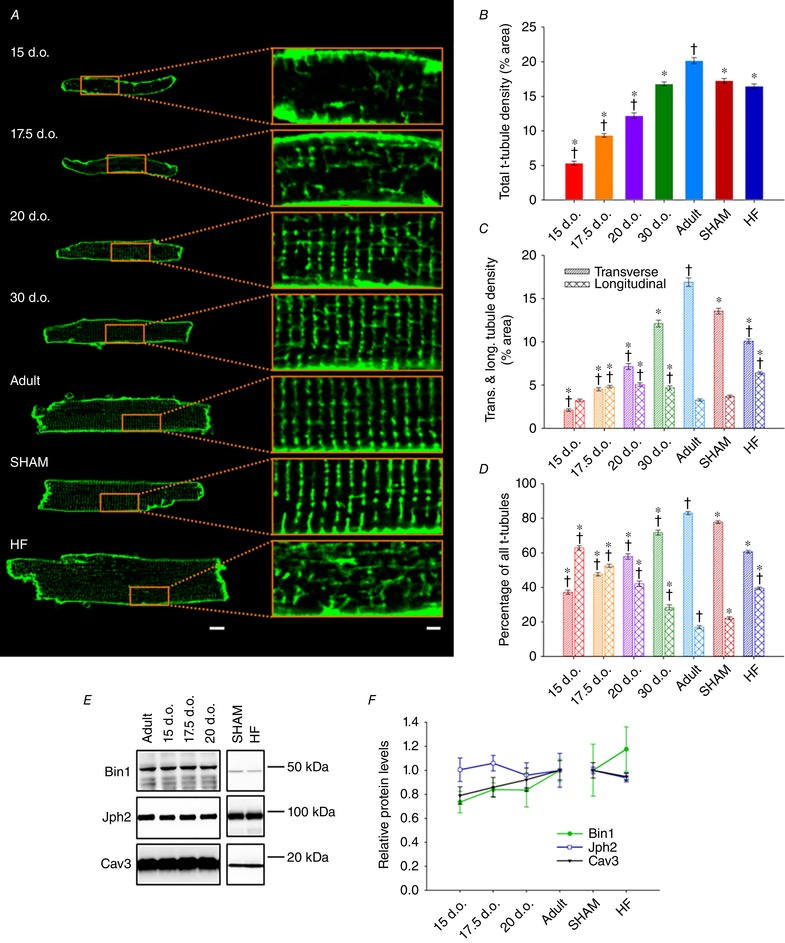
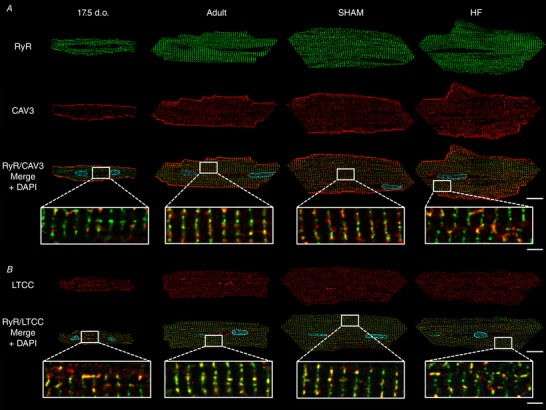
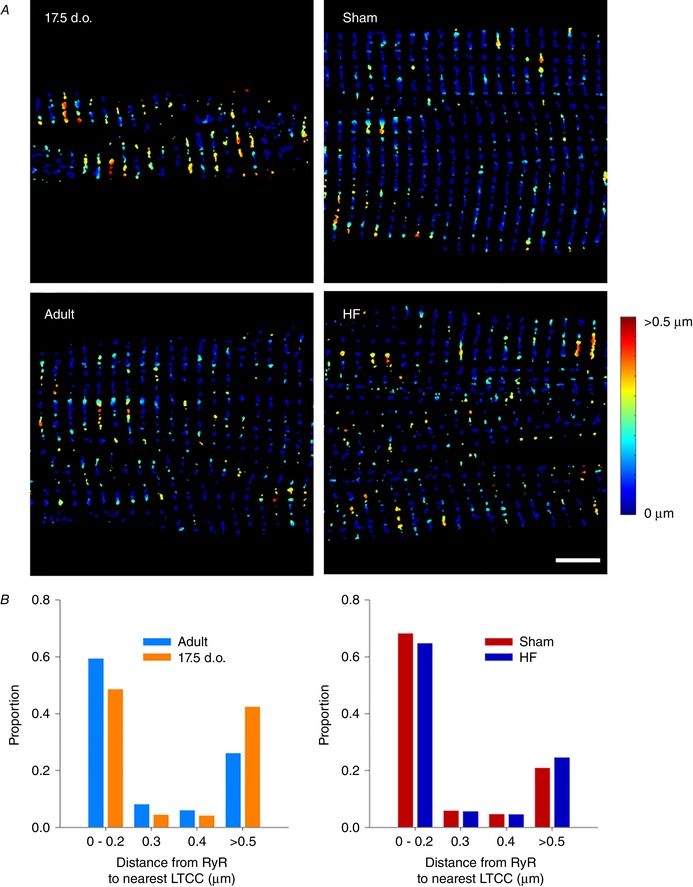
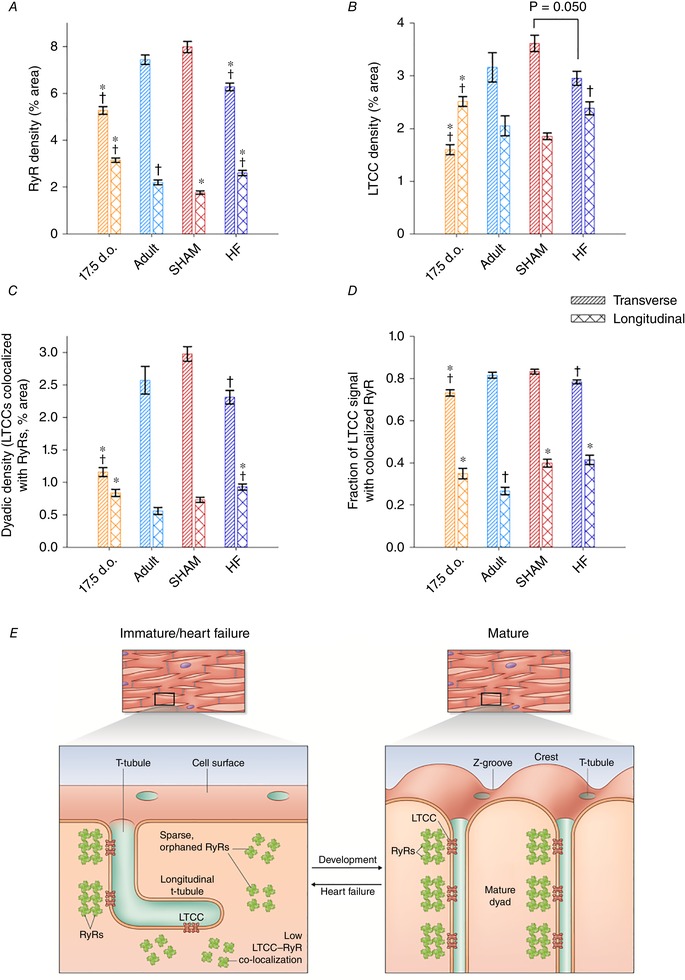
Reactivation of the fetal gene programme has been implicated as a driver of pathological cardiac remodelling. Here we examined whether pathological remodelling of cardiomyocyte substructure and function during heart failure (HF) reflects a reversion to an immature phenotype. Using scanning electron microscopy, we observed that Z-grooves and t-tubule openings at the cell surface appeared gradually during cardiac development, and disappeared during HF. Confocal and super-resolution imaging within the cell interior revealed similar structural parallels; disorganization of t-tubules in failing cells was strikingly reminiscent of the late stages of postnatal development, with fewer transverse elements and a high proportion of longitudinal tubules. Ryanodine receptors (RyRs) were observed to be laid down in advance of developing t-tubules and similarly 'orphaned' in HF, although RyR distribution along Z-lines was relatively sparse. Indeed, nanoscale imaging revealed coordinated packing of L-type Ca channels and RyRs into dyadic junctions during development, and orderly unpacking during HF. These findings support a 'last in, first out' paradigm, as the latest stages of dyadic structural development are reversed during disease. Paired imaging of t-tubules and Ca showed that the disorganized arrangement of dyads in immature and failing cells promoted desynchronized and slowed Ca release in these two states. However, while developing cells exhibited efficient triggering of Ca release at newly formed dyads, dyadic function was impaired in failing cells despite similar organization of Ca handling proteins. Thus, pathologically deficient Ca homeostasis during HF is only partly linked to the re-emergence of immature subcellular structure, and additionally reflects lost dyadic functionality.
胎儿基因程序的再激活已被认为与心力衰竭(HF)期间的病理性重塑有关,因此我们研究了在这种疾病过程中心肌细胞亚细胞结构和功能是否恢复到未成熟表型。表面和内部膜结构在发育过程中逐渐出现,并在 HF 期间恢复到幼年状态。同样,在发育过程中,细胞膜和肌浆网之间的二联体连接逐渐用 L 型 Ca 通道和兰尼碱受体“包装”,而在 HF 期间则“拆开”。尽管亚细胞结构相似,但在早期发育阶段观察到二联体是功能性的,但在衰竭的心肌细胞中,Ca 释放能力受损。因此,尽管未成熟和衰竭的心肌细胞在亚细胞结构上存在相似之处,但这并不能完全解释 HF 中观察到的 Ca 稳态的显著损害。
胎儿基因程序的再激活已被认为是病理性心脏重塑的驱动因素。在这里,我们研究了心力衰竭(HF)期间心肌细胞亚结构和功能的病理性重塑是否反映了向未成熟表型的逆转。我们使用扫描电子显微镜观察到,在心脏发育过程中,Z 槽和 T 管开口逐渐出现在细胞表面,而在 HF 期间则消失。细胞内部的共聚焦和超分辨率成像显示出类似的结构平行;在衰竭细胞中 T 管的紊乱排列令人联想到出生后发育的晚期,横向元素较少,纵向管较多。尽管 RyR 分布在 Z 线上相对稀疏,但在 T 管发育之前观察到 RyR 的沉积,并在 HF 中同样“孤立”。实际上,纳米级成像显示 L 型 Ca 通道和 RyR 在发育过程中协调地包装成二联体连接,并在 HF 期间有序地拆开。这些发现支持“最后进来,首先出去”的范例,因为疾病期间会逆转二联体结构发育的最新阶段。T 管和 Ca 的配对成像表明,在未成熟和衰竭细胞中二联体的紊乱排列促进了这两种状态下 Ca 释放的不同步和减缓。然而,尽管发育中的细胞在新形成的二联体中表现出有效的 Ca 释放触发,但在衰竭细胞中二联体功能受损,尽管 Ca 处理蛋白的组织相似。因此,HF 期间病理性 Ca 稳态不足部分与未成熟亚细胞结构的重新出现有关,此外还反映了丢失的二联体功能。