CAS Key Lab of Computational Biology, CAS-MPG Partner Institute for Computational Biology, Shanghai Institute of Nutrition and Health, Shanghai Institute for Biological Sciences, University of Chinese Academy of Sciences, Chinese Academy of Sciences, Shanghai, China.
UCL Cancer Institute, University College London, London, UK.
Bioinformatics. 2019 Sep 15;35(18):3514-3516. doi: 10.1093/bioinformatics/btz073.
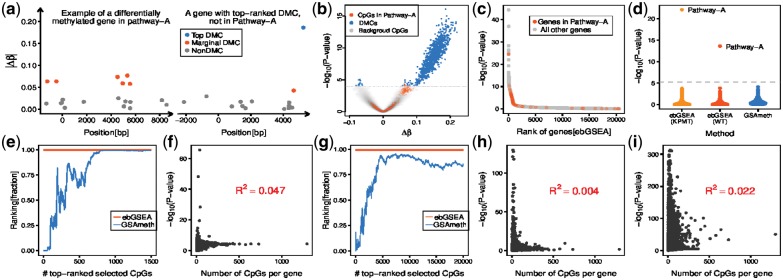
The biological interpretation of differentially methylated sites derived from Epigenome-Wide-Association Studies (EWAS) remains a significant challenge. Gene Set Enrichment Analysis (GSEA) is a general tool to aid biological interpretation, yet its correct and unbiased implementation in the EWAS context is difficult due to the differential probe representation of Illumina Infinium DNA methylation beadchips.
We present a novel GSEA method, called ebGSEA, which ranks genes, not CpGs, according to the overall level of differential methylation, as assessed using all the probes mapping to the given gene. Applied on simulated and real EWAS data, we show how ebGSEA may exhibit higher sensitivity and specificity than the current state-of-the-art, whilst also avoiding differential probe representation bias. Thus, ebGSEA will be a useful additional tool to aid the interpretation of EWAS data.
ebGSEA is available from https://github.com/aet21/ebGSEA, and has been incorporated into the ChAMP Bioconductor package (https://www.bioconductor.org).
Supplementary data are available at Bioinformatics online.
从表观基因组关联研究(EWAS)中得出的差异甲基化位点的生物学解释仍然是一个重大挑战。基因集富集分析(GSEA)是一种辅助生物学解释的通用工具,但由于 Illumina Infinium DNA 甲基化 beadchips 对探针的差异表示,正确且无偏地在 EWAS 环境中实施 GSEA 具有一定难度。
我们提出了一种新的 GSEA 方法,称为 ebGSEA,它根据使用映射到给定基因的所有探针评估的整体差异甲基化水平对基因进行排序,而不是对 CpG 进行排序。在模拟和真实的 EWAS 数据上的应用表明,与当前最先进的方法相比,ebGSEA 可能具有更高的灵敏度和特异性,同时避免了差异探针表示偏差。因此,ebGSEA 将是辅助 EWAS 数据解释的有用辅助工具。
ebGSEA 可从 https://github.com/aet21/ebGSEA 获得,并已被纳入 ChAMP Bioconductor 包(https://www.bioconductor.org)。
补充数据可在 Bioinformatics 在线获得。