Division of Clinical and Metabolic Genetics, The Hospital for Sick Children, Toronto, ON, Canada.
Medical Genetics Residency Training Program, University of Toronto, Toronto, ON, Canada.
J Neurodev Disord. 2019 Feb 7;11(1):3. doi: 10.1186/s11689-019-9263-3.
Ultra-rare genetic variants, including non-recurrent copy number variations (CNVs) affecting important dosage-sensitive genes, are important contributors to the etiology of neurodevelopmental disorders (NDDs). Pairing family-based whole-genome sequencing (WGS) with detailed phenotype data can enable novel gene associations in NDDs.
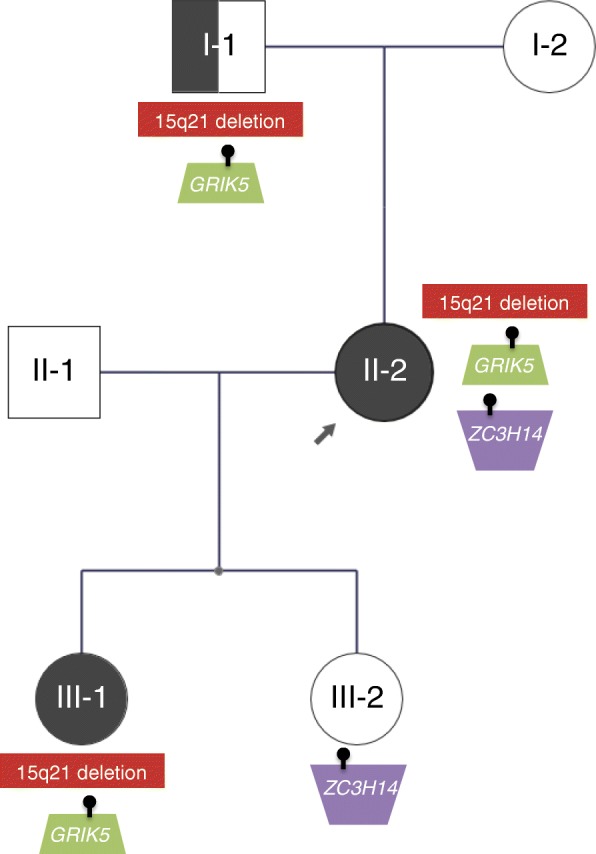
We performed WGS of six members from a three-generation family, where three individuals each had a spectrum of features suggestive of a NDD. CNVs and sequence-level variants were identified and further investigated in disease and control databases.
We identified a novel 252-kb deletion at 15q21 that overlaps the synaptic gene DMXL2 and the gene GLDN. The microdeletion segregated in NDD-affected individuals. Additional rare inherited and de novo sequence-level variants were found that may also be involved, including a missense change in GRIK5. Multiple CNVs and loss-of-function sequence variants affecting DMXL2 were discovered in additional unrelated individuals with a range of NDDs.
Disruption of DMXL2 may predispose to NDDs including autism spectrum disorder. The robust interpretation of private variants requires a multifaceted approach that incorporates multigenerational pedigrees and genome-wide and population-scale data.
超罕见的遗传变异,包括影响重要剂量敏感基因的非重复拷贝数变异(CNV),是神经发育障碍(NDD)病因学的重要贡献者。将基于家族的全基因组测序(WGS)与详细的表型数据相结合,可以在 NDD 中发现新的基因关联。
我们对一个三代家族的六名成员进行了 WGS,其中三个人各自具有一系列提示 NDD 的特征。在疾病和对照数据库中鉴定和进一步研究了 CNV 和序列水平变体。
我们在 15q21 上发现了一个新的 252kb 缺失,该缺失重叠了突触基因 DMXL2 和 GLDN。微缺失在受 NDD 影响的个体中分离。还发现了其他罕见的遗传性和新生序列水平变体可能也参与其中,包括 GRIK5 的错义变化。在患有各种 NDD 的其他无关个体中发现了多个影响 DMXL2 的 CNV 和功能丧失序列变体。
DMXL2 的破坏可能易患包括自闭症谱系障碍在内的 NDD。对私人变体的有力解释需要采用多方面的方法,包括多代系谱以及全基因组和人群规模的数据。