Dr. John T. Macdonald Foundation Department of Human Genetics, John P. Hussman Institute for Human Genomics, University of Miami Miller School of Medicine, Miami, Florida, USA.
Department of Computer Science and Department of Biology, University of Miami, Coral Gables, Florida, USA.
Sci Rep. 2019 Feb 8;9(1):1692. doi: 10.1038/s41598-018-37119-z.
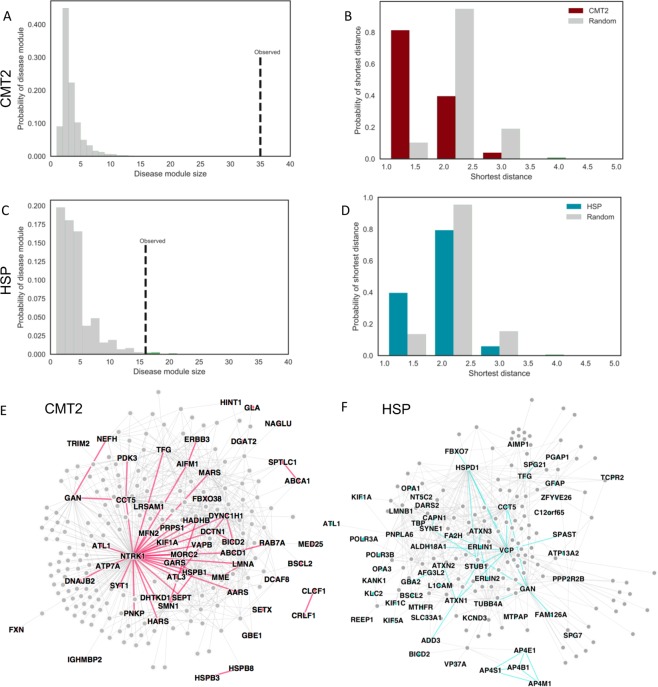
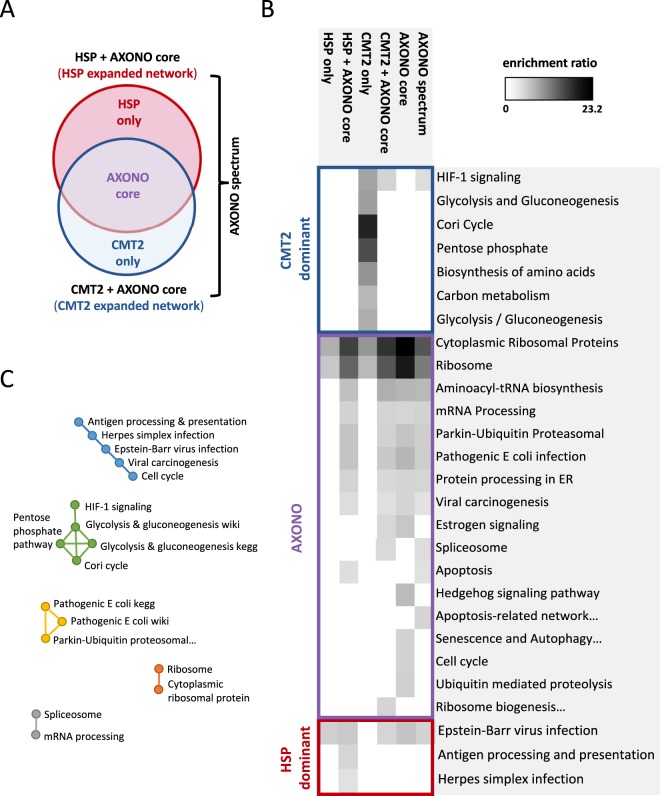
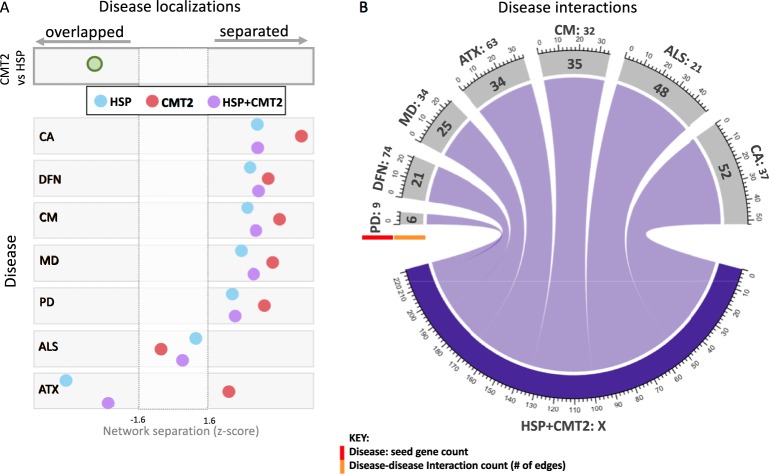
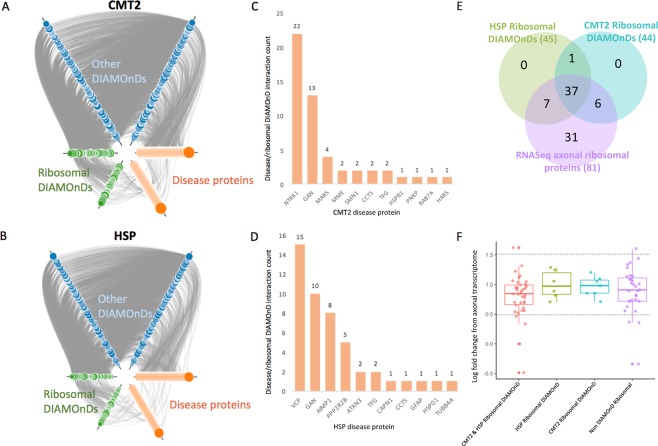
Inherited axonopathies represent a spectrum of disorders unified by the common pathological mechanism of length-dependent axonal degeneration. Progressive axonal degeneration can lead to both Charcot-Marie-Tooth type 2 (CMT2) and Hereditary Spastic Paraplegia (HSP) depending on the affected neurons: peripheral motor and sensory nerves or central nervous system axons of the corticospinal tract and dorsal columns, respectively. Inherited axonopathies display an extreme degree of genetic heterogeneity of Mendelian high-penetrance genes. High locus heterogeneity is potentially advantageous to deciphering disease etiology by providing avenues to explore biological pathways in an unbiased fashion. Here, we investigate 'gene modules' in inherited axonopathies through a network-based analysis of the Human Integrated Protein-Protein Interaction rEference (HIPPIE) database. We demonstrate that CMT2 and HSP disease proteins are significantly more connected than randomly expected. We define these connected disease proteins as 'proto-modules' and show the topological relationship of these proto-modules by evaluating their overlap through a shortest-path based measurement. In particular, we observe that the CMT2 and HSP proto-modules significantly overlapped, demonstrating a shared genetic etiology. Comparison of both modules with other diseases revealed an overlapping relationship between HSP and hereditary ataxia and between CMT2 + HSP and hereditary ataxia. We then use the DIseAse Module Detection (DIAMOnD) algorithm to expand the proto-modules into comprehensive disease modules. Analysis of disease modules thus obtained reveals an enrichment of ribosomal proteins and pathways likely central to inherited axonopathy pathogenesis, including protein processing in the endoplasmic reticulum, spliceosome, and mRNA processing. Furthermore, we determine pathways specific to each axonopathy by analyzing the difference of the axonopathy modules. CMT2-specific pathways include glycolysis and gluconeogenesis-related processes, while HSP-specific pathways include processes involved in viral infection response. Unbiased characterization of inherited axonopathy disease modules will provide novel candidate disease genes, improve interpretation of candidate genes identified through patient data, and guide therapy development.
遗传性轴突病代表了一组疾病,它们具有共同的长度依赖性轴突变性病理机制,因此具有统一性。进行性轴突变性可导致夏科-马里-图雷特病 2 型(CMT2)和遗传性痉挛性截瘫(HSP),这取决于受影响的神经元:外周运动和感觉神经或皮质脊髓束和背柱的中枢神经系统轴突。遗传性轴突病表现出孟德尔高外显率基因的遗传异质性的极端程度。高基因座异质性对于通过提供以无偏倚的方式探索生物学途径的途径来解析疾病病因具有潜在优势。在这里,我们通过对人类综合蛋白质-蛋白质相互作用参考(HIPPIE)数据库进行基于网络的分析,研究遗传性轴突病中的“基因模块”。我们证明 CMT2 和 HSP 疾病蛋白比随机预期的连接更多。我们将这些连接的疾病蛋白定义为“原模块”,并通过基于最短路径的测量来评估它们的重叠,从而显示这些原模块的拓扑关系。特别是,我们观察到 CMT2 和 HSP 原模块显著重叠,表明存在共同的遗传病因。将两个模块与其他疾病进行比较发现,HSP 与遗传性共济失调之间以及 CMT2+HSP 与遗传性共济失调之间存在重叠关系。然后,我们使用疾病模块检测(DIAMOnD)算法将原模块扩展为综合疾病模块。对由此获得的疾病模块进行分析表明,核糖体蛋白和可能对遗传性轴突病发病机制至关重要的途径富集,包括内质网、剪接体和 mRNA 加工中的蛋白质加工。此外,我们通过分析轴突病模块的差异来确定每个轴突病特有的途径。CMT2 特有的途径包括糖酵解和糖异生相关过程,而 HSP 特有的途径包括涉及病毒感染反应的过程。对遗传性轴突病疾病模块进行无偏特征描述将提供新的候选疾病基因,改善通过患者数据鉴定的候选基因的解释,并指导治疗的发展。