Department of Computing, National University of Computer and Emerging Sciences, Islamabad, 40100, Pakistan.
Computational Biology Research Lab, Islamabad, 40100, Pakistan.
BMC Bioinformatics. 2020 Nov 4;21(1):500. doi: 10.1186/s12859-020-03827-5.
High throughput experiments have generated a significantly large amount of protein interaction data, which is being used to study protein networks. Studying complete protein networks can reveal more insight about healthy/disease states than studying proteins in isolation. Similarly, a comparative study of protein-protein interaction (PPI) networks of different species reveals important insights which may help in disease analysis and drug design. The study of PPI network alignment can also helps in understanding the different biological systems of different species. It can also be used in transfer of knowledge across different species. Different aligners have been introduced in the last decade but developing an accurate and scalable global alignment algorithm that can ensures the biological significance alignment is still challenging.
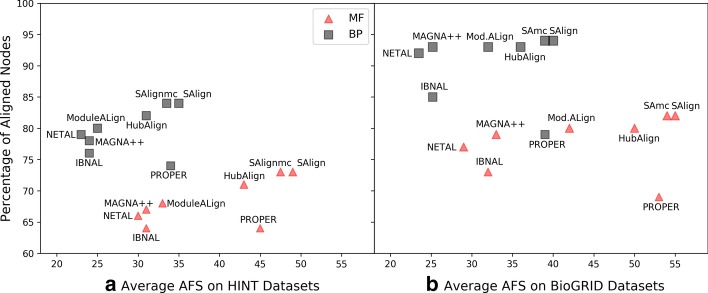
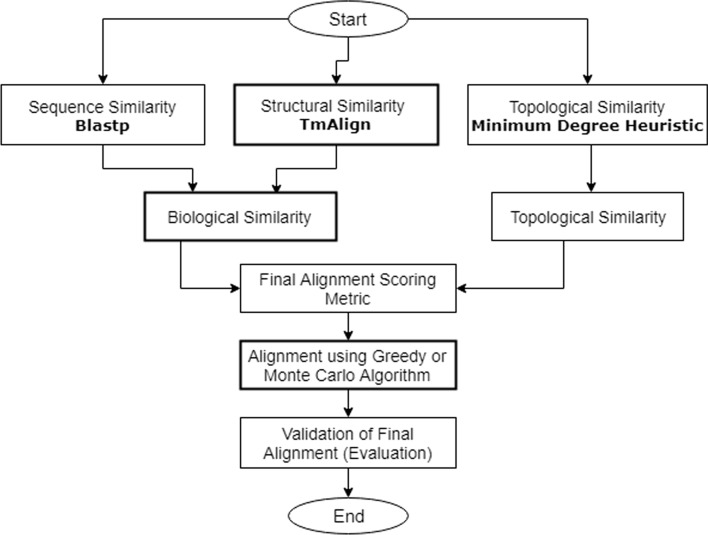
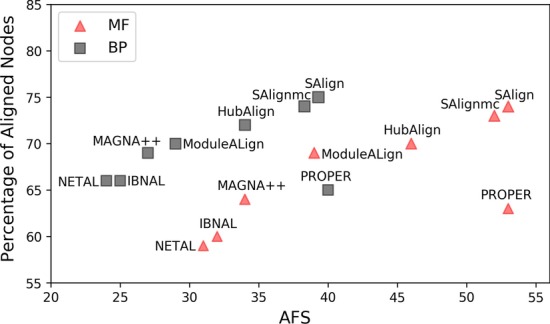
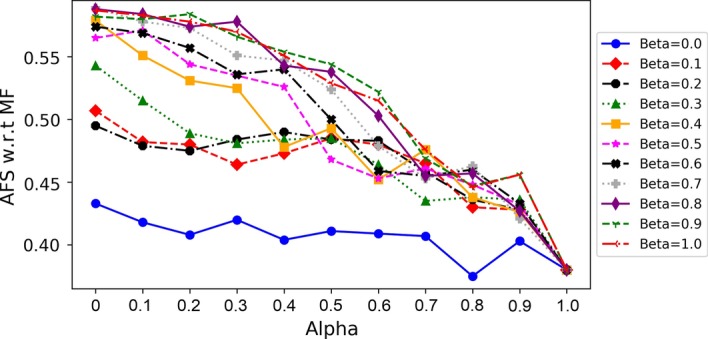
This paper presents a novel global pairwise network alignment algorithm, SAlign, which uses topological and biological information in the alignment process. The proposed algorithm incorporates sequence and structural information for computing biological scores, whereas previous algorithms only use sequence information. The alignment based on the proposed technique shows that the combined effect of structure and sequence results in significantly better pairwise alignments. We have compared SAlign with state-of-art algorithms on the basis of semantic similarity of alignment and the number of aligned nodes on multiple PPI network pairs. The results of SAlign on the network pairs which have high percentage of proteins with available structure are 3-63% semantically better than all existing techniques. Furthermore, it also aligns 5-14% more nodes of these network pairs as compared to existing aligners. The results of SAlign on other PPI network pairs are comparable or better than all existing techniques. We also introduce [Formula: see text], a Monte Carlo based alignment algorithm, that produces multiple network alignments with similar semantic similarity. This helps the user to pick biologically meaningful alignments.
The proposed algorithm has the ability to find the alignments that are more biologically significant/relevant as compared to the alignments of existing aligners. Furthermore, the proposed method is able to generate alternate alignments that help in studying different genes/proteins of the specie.
高通量实验产生了大量的蛋白质相互作用数据,这些数据被用于研究蛋白质网络。研究完整的蛋白质网络比单独研究蛋白质能揭示更多关于健康/疾病状态的信息。同样,对不同物种的蛋白质-蛋白质相互作用(PPI)网络进行比较研究,可以揭示重要的见解,有助于疾病分析和药物设计。研究 PPI 网络比对还有助于理解不同物种的不同生物系统。它还可以用于在不同物种之间转移知识。过去十年中已经引入了不同的比对器,但开发一种能够确保生物意义比对的准确和可扩展的全局比对算法仍然具有挑战性。
本文提出了一种新的全局成对网络比对算法 SAlign,该算法在比对过程中使用拓扑和生物学信息。所提出的算法将序列和结构信息用于计算生物学得分,而以前的算法仅使用序列信息。基于所提出的技术的比对表明,结构和序列的综合效果导致了显著更好的成对比对。我们已经基于语义相似性和多个 PPI 网络对的对齐节点数,将 SAlign 与最先进的算法进行了比较。SAlign 在具有大量可用结构的蛋白质的网络对上的结果在语义上比所有现有技术好 3-63%。此外,与现有比对器相比,它还对齐了这些网络对中的 5-14%更多的节点。SAlign 在其他 PPI 网络对上的结果与所有现有技术相当或更好。我们还引入了基于蒙特卡罗的比对算法 [Formula: see text],该算法可以生成具有相似语义相似性的多个网络比对。这有助于用户选择具有生物学意义的比对。
与现有比对器的比对相比,所提出的算法具有发现更具生物学意义/相关性的比对的能力。此外,所提出的方法能够生成有助于研究特定物种不同基因/蛋白质的替代比对。