Dr. John T. Macdonald Foundation Department of Human Genetics, John P. Hussman Institute for Human Genomics, University of Miami Miller School of Medicine, Miami, FL, USA.
Department of Human Genetics, McGill University, Montréal, QC, Canada.
Genet Med. 2020 Dec;22(12):2114-2119. doi: 10.1038/s41436-020-0924-0. Epub 2020 Aug 3.
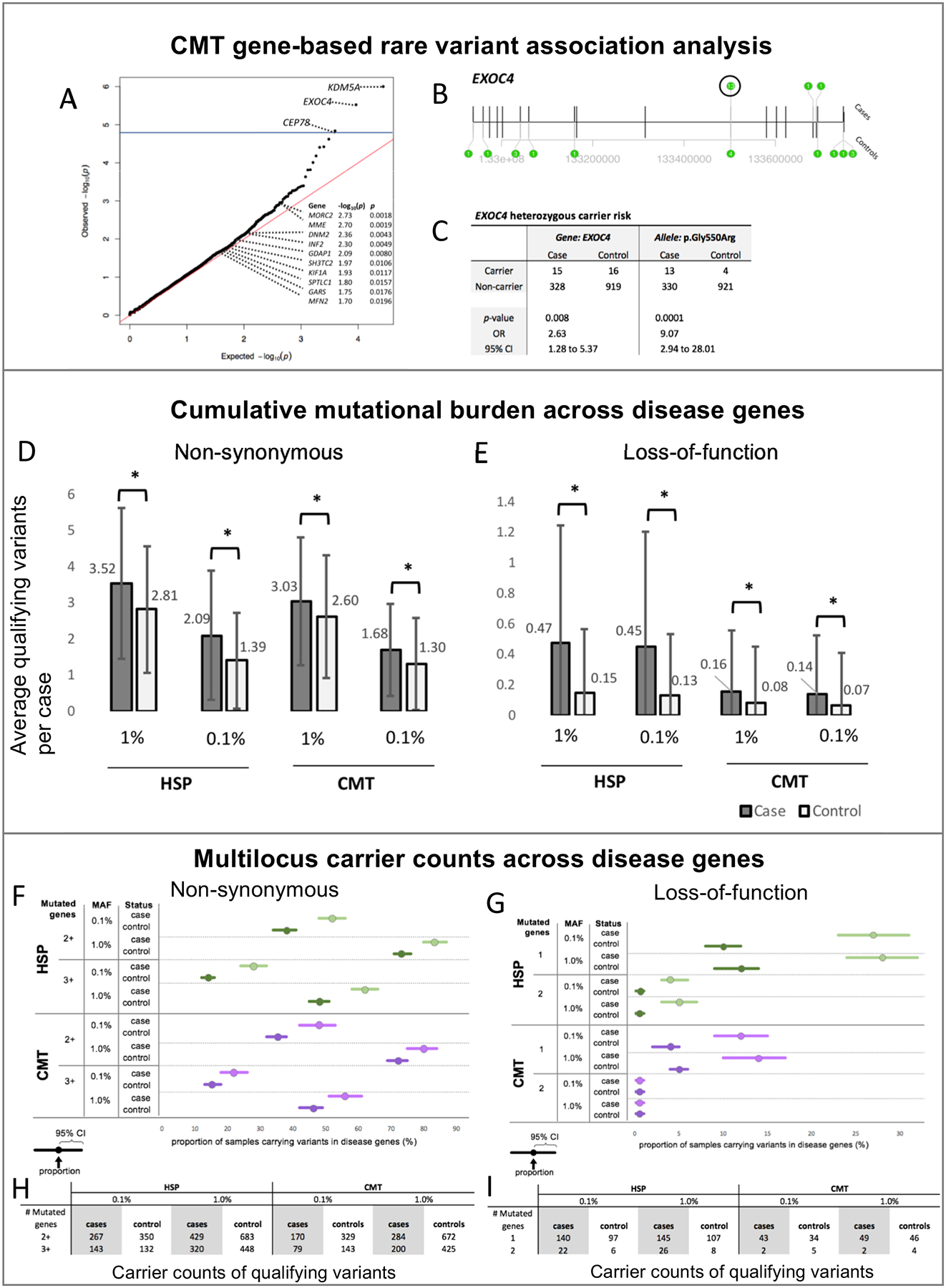
Inherited axonopathies (IA) are rare, clinically and genetically heterogeneous diseases that lead to length-dependent degeneration of the long axons in central (hereditary spastic paraplegia [HSP]) and peripheral (Charcot-Marie-Tooth type 2 [CMT2]) nervous systems. Mendelian high-penetrance alleles in over 100 different genes have been shown to cause IA; however, about 50% of IA cases do not receive a genetic diagnosis. A more comprehensive spectrum of causative genes and alleles is warranted, including causative and risk alleles, as well as oligogenic multilocus inheritance.
Through international collaboration, IA exome studies are beginning to be sufficiently powered to perform a pilot rare variant burden analysis. After extensive quality control, our cohort contained 343 CMT cases, 515 HSP cases, and 935 non-neurological controls. We assessed the cumulative mutational burden across disease genes, explored the evidence for multilocus inheritance, and performed an exome-wide rare variant burden analysis.
We replicated the previously described mutational burden in a much larger cohort of CMT cases, and observed the same effect in HSP cases. We identified a preliminary risk allele for CMT in the EXOC4 gene (p value= 6.9 × 10-6, odds ratio [OR] = 2.1) and explored the possibility of multilocus inheritance in IA.
Our results support the continuing emergence of complex inheritance mechanisms in historically Mendelian disorders.
遗传性轴索神经病(IA)是罕见的、临床表现和遗传异质性的疾病,导致中枢神经系统(遗传性痉挛性截瘫[HSP])和周围神经系统(Charcot-Marie-Tooth 型 2 [CMT2])中的长轴突进行性退化。已经证实超过 100 个不同基因中的孟德尔高外显率等位基因可导致 IA;然而,约 50%的 IA 病例未得到遗传诊断。因此,需要更全面的致病基因和等位基因谱,包括致病和风险等位基因,以及多基因多部位遗传。
通过国际合作,IA 外显子组研究开始具有足够的效力来进行罕见变异负担分析的试点研究。经过广泛的质量控制,我们的队列包含 343 例 CMT 病例、515 例 HSP 病例和 935 例非神经科对照。我们评估了疾病基因的累积突变负担,探索了多基因遗传的证据,并进行了外显子组范围内的罕见变异负担分析。
我们在更大的 CMT 病例队列中复制了先前描述的突变负担,并在 HSP 病例中观察到了相同的影响。我们在 EXOC4 基因中确定了一个 CMT 的初步风险等位基因(p 值=6.9×10-6,优势比[OR] =2.1),并探索了 IA 中多基因遗传的可能性。
我们的结果支持在历史上孟德尔疾病中不断出现复杂的遗传机制。