Laboratory Services Section, Department of Transfusion Medicine, NIH Clinical Center, National Institutes of Health, Bethesda, MD, 20892, USA.
Bioinformatics and Computational Biosciences Branch, Office of Cyber Infrastructure and Computational Biology, National Institute of Allergy and Infectious Diseases, Bethesda, MD, USA.
J Transl Med. 2019 Feb 11;17(1):43. doi: 10.1186/s12967-019-1791-9.
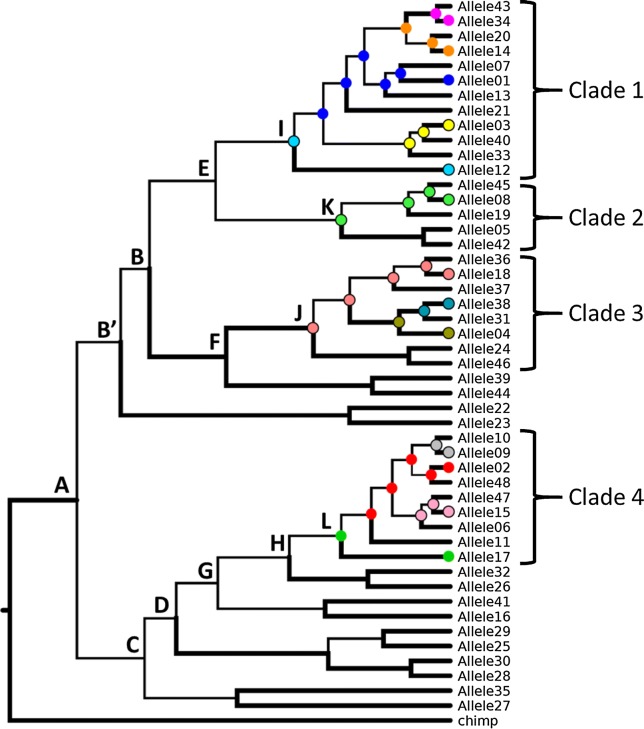
Sequence information generated from next generation sequencing is often computationally phased using haplotype-phasing algorithms. Utilizing experimentally derived allele or haplotype information improves this prediction, as routinely used in HLA typing. We recently established a large dataset of long ERMAP alleles, which code for protein variants in the Scianna blood group system. We propose the phylogeny of this set of 48 alleles and identify evolutionary steps to derive the observed alleles.
The nucleotide sequence of > 21 kb each was used for all physically confirmed 48 ERMAP alleles that we previously published. Full-length sequences were aligned and variant sites were extracted manually. The Bayesian coalescent algorithm implemented in BEAST v1.8.3 was used to estimate a coalescent phylogeny for these variants and the allelic ancestral states at the internal nodes of the phylogeny.
The phylogenetic analysis allowed us to identify the evolutionary relationships among the 48 ERMAP alleles, predict 4243 potential ancestral alleles and calculate a posterior probability for each of these unobserved alleles. Some of them coincide with observed alleles that are extant in the population.
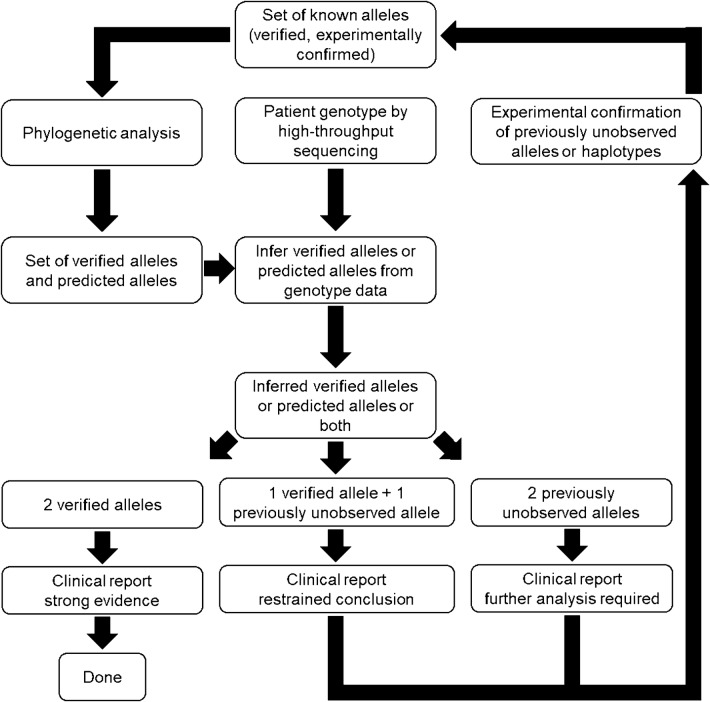
Our proposed strategy places known alleles in a phylogenetic framework, allowing us to describe as-yet-undiscovered alleles. In this new approach, which relies heavily on the accuracy of the alleles used for the phylogenetic analysis, an expanded set of predicted alleles can be used to infer alleles when large genotype data are analyzed, as typically generated by high-throughput sequencing. The alleles identified by studies like ours may be utilized in designing of microarray technologies, imputing of genotypes and mapping of next generation sequencing data.
下一代测序产生的序列信息通常使用单倍型相位算法进行计算相位。利用实验得出的等位基因或单倍型信息可以改善这种预测,这在 HLA 分型中经常使用。我们最近建立了一个包含大量长 ERMAP 等位基因的数据集,这些等位基因编码 Scianna 血型系统中的蛋白质变体。我们提出了这组 48 个等位基因的系统发育,并确定了推导观察到的等位基因的进化步骤。
我们之前发表的所有经过物理确认的 48 个 ERMAP 等位基因,每个等位基因的核苷酸序列都超过 21kb。使用全序列进行比对,并手动提取变异位点。贝叶斯合并算法(在 BEAST v1.8.3 中实现)用于估计这些变体的合并系统发育,以及系统发育内部节点的等位基因祖先状态。
系统发育分析使我们能够确定 48 个 ERMAP 等位基因之间的进化关系,预测 4243 个潜在的祖先等位基因,并为每个未观察到的等位基因计算后验概率。其中一些与现存于人群中的观察到的等位基因吻合。
我们提出的策略将已知的等位基因置于系统发育框架中,使我们能够描述尚未发现的等位基因。在这种新方法中,严重依赖于用于系统发育分析的等位基因的准确性,可以在分析大型基因型数据时使用扩展的预测等位基因集来推断等位基因,如高通量测序通常产生的。像我们这样的研究中确定的等位基因可以用于设计微阵列技术、基因型推断和下一代测序数据的映射。