Liu Yu, Koyutürk Mehmet, Maxwell Sean, Xiang Min, Veigl Martina, Cooper Richard S, Tayo Bamidele O, Li Li, LaFramboise Thomas, Wang Zhenghe, Zhu Xiaofeng, Chance Mark R
Center for Proteomics and Bioinformatics, Case Western Reserve University, Cleveland, OH, USA.
BMC Genomics. 2014 Aug 16;15(1):685. doi: 10.1186/1471-2164-15-685.
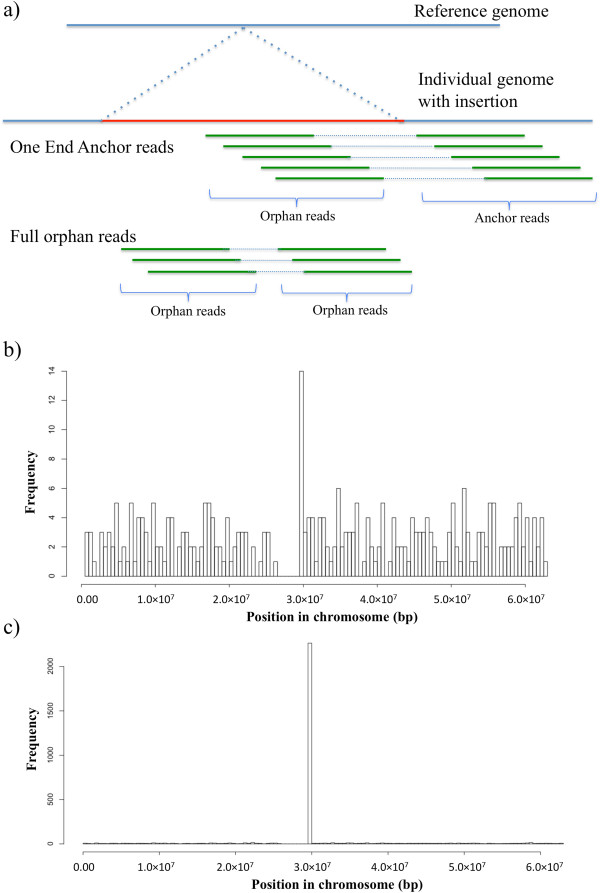
Sequences up to several megabases in length have been found to be present in individual genomes but absent in the human reference genome. These sequences may be common in populations, and their absence in the reference genome may indicate rare variants in the genomes of individuals who served as donors for the human genome project. As the reference genome is used in probe design for microarray technology and mapping short reads in next generation sequencing (NGS), this missing sequence could be a source of bias in functional genomic studies and variant analysis. One End Anchor (OEA) and/or orphan reads from paired-end sequencing have been used to identify novel sequences that are absent in reference genome. However, there is no study to investigate the distribution, evolution and functionality of those sequences in human populations.
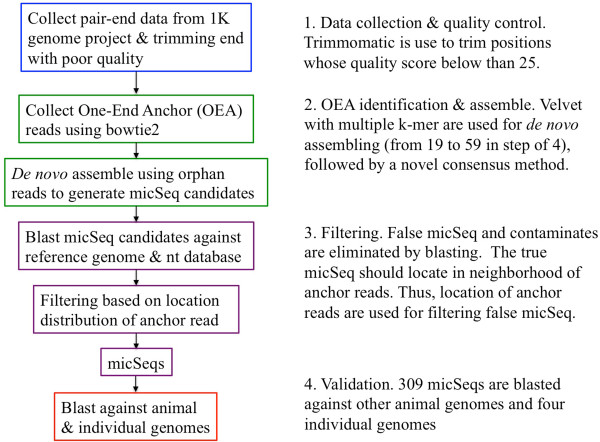
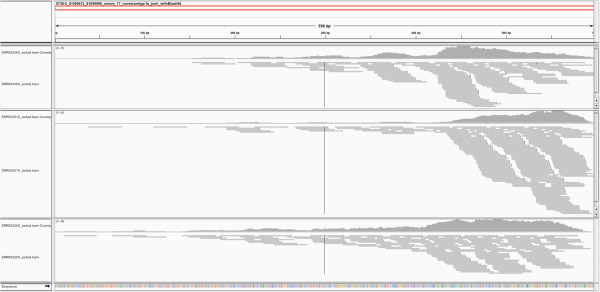
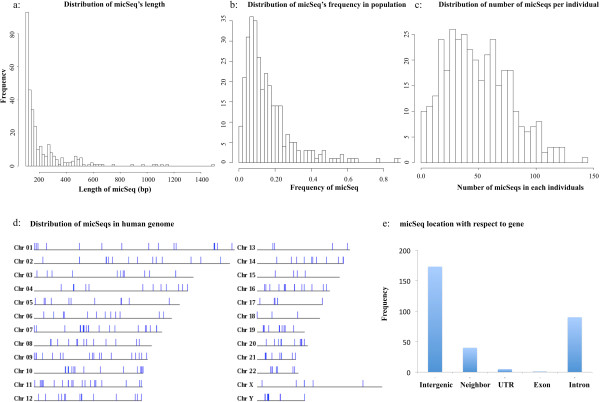
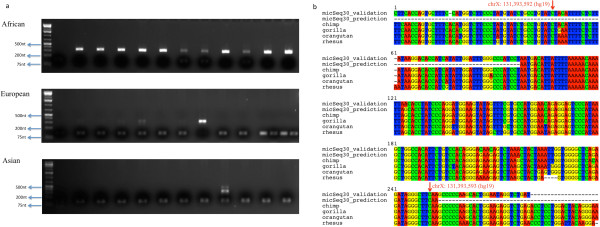
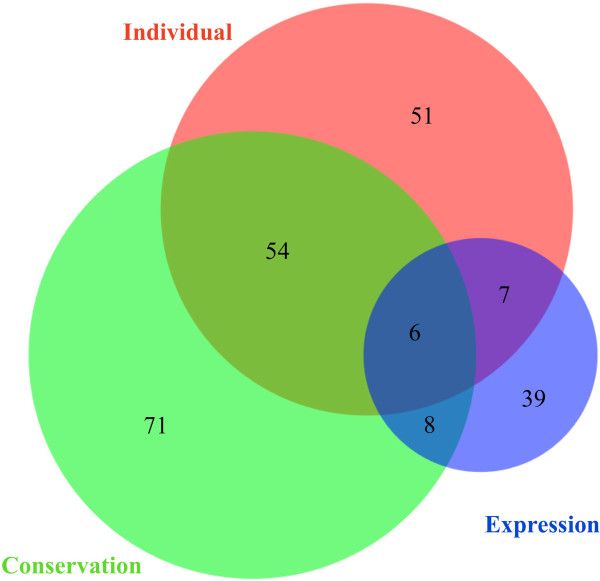
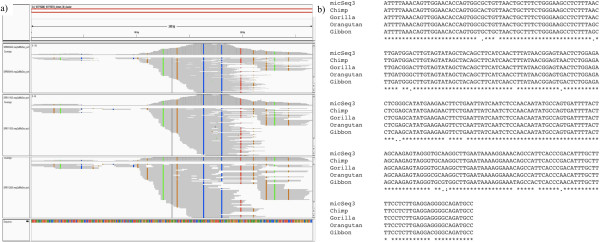
To systematically identify and study the missing common sequences (micSeqs), we extended the previous method by pooling OEA reads from large number of individuals and applying strict filtering methods to remove false sequences. The pipeline was applied to data from phase 1 of the 1000 Genomes Project. We identified 309 micSeqs that are present in at least 1% of the human population, but absent in the reference genome. We confirmed 76% of these 309 micSeqs by comparison to other primate genomes, individual human genomes, and gene expression data. Furthermore, we randomly selected fifteen micSeqs and confirmed their presence using PCR validation in 38 additional individuals. Functional analysis using published RNA-seq and ChIP-seq data showed that eleven micSeqs are highly expressed in human brain and three micSeqs contain transcription factor (TF) binding regions, suggesting they are functional elements. In addition, the identified micSeqs are absent in non-primates and show dynamic acquisition during primate evolution culminating with most micSeqs being present in Africans, suggesting some micSeqs may be important sources of human diversity.
76% of micSeqs were confirmed by a comparative genomics approach. Fourteen micSeqs are expressed in human brain or contain TF binding regions. Some micSeqs are primate-specific, conserved and may play a role in the evolution of primates.
已发现个体基因组中存在长达数兆碱基的序列,但在人类参考基因组中却不存在。这些序列在人群中可能很常见,而它们在参考基因组中的缺失可能表明作为人类基因组计划供体的个体基因组中存在罕见变异。由于参考基因组用于微阵列技术的探针设计以及下一代测序(NGS)中的短读段映射,这种缺失序列可能是功能基因组研究和变异分析中偏差的一个来源。单端锚定(OEA)和/或双端测序中的孤儿读段已被用于识别参考基因组中不存在的新序列。然而,尚无研究调查这些序列在人群中的分布、进化和功能。
为了系统地识别和研究缺失的常见序列(micSeqs),我们扩展了先前的方法,通过汇集大量个体的OEA读段并应用严格的过滤方法来去除假序列。该流程应用于千人基因组计划第一阶段的数据。我们鉴定出309个micSeqs,它们存在于至少1%的人类群体中,但在参考基因组中不存在。通过与其他灵长类基因组、个体人类基因组和基因表达数据进行比较,我们证实了这309个micSeqs中的76%。此外,我们随机选择了15个micSeqs,并在另外38个个体中使用PCR验证确认了它们的存在。使用已发表的RNA-seq和ChIP-seq数据进行的功能分析表明,11个micSeqs在人类大脑中高度表达,3个micSeqs包含转录因子(TF)结合区域,表明它们是功能元件。此外,鉴定出的micSeqs在非灵长类动物中不存在,并在灵长类进化过程中呈现动态获得,最终大多数micSeqs存在于非洲人群中,这表明一些micSeqs可能是人类多样性的重要来源。
通过比较基因组学方法证实了76%的micSeqs。14个micSeqs在人类大脑中表达或包含TF结合区域。一些micSeqs是灵长类特有的、保守的,可能在灵长类进化中发挥作用。