Hagihara Hideo, Ohira Koji, Miyakawa Tsuyoshi
Division of Systems Medical Science, Institute for Comprehensive Medical Science, Fujita Health University, Toyoake, Japan.
Laboratory of Nutritional Brain Science, Department of Food Science and Nutrition, Mukogawa Women's University, Nishinomiya, Japan.
Neuropsychopharmacol Rep. 2019 Jun;39(2):78-89. doi: 10.1002/npr2.12048. Epub 2019 Feb 16.
The molecular and cellular mechanisms underlying the antidepressant effects of fluoxetine in the brain are not fully understood. Emerging evidence has led to the hypothesis that chronic fluoxetine treatment induces dematuration of certain types of mature neurons in rodents. These studies have focused on the properties of typical molecular and/or electrophysiological markers for neuronal maturation. Nevertheless, it remains unknown whether dematuration-related phenomena are present at the genome-wide gene expression level.
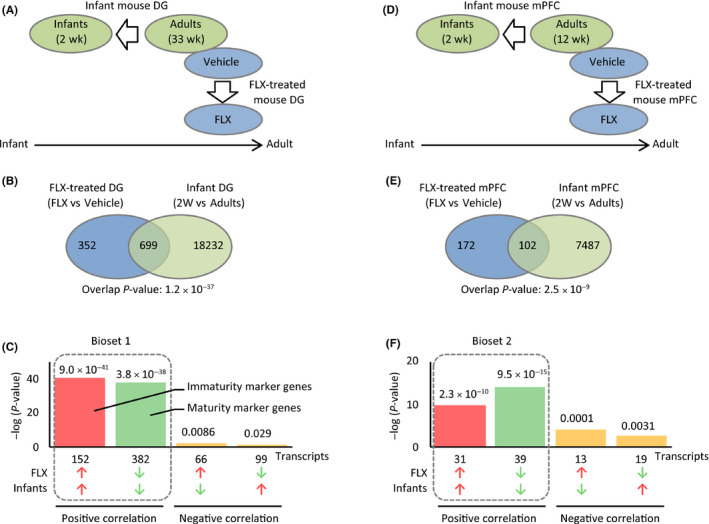
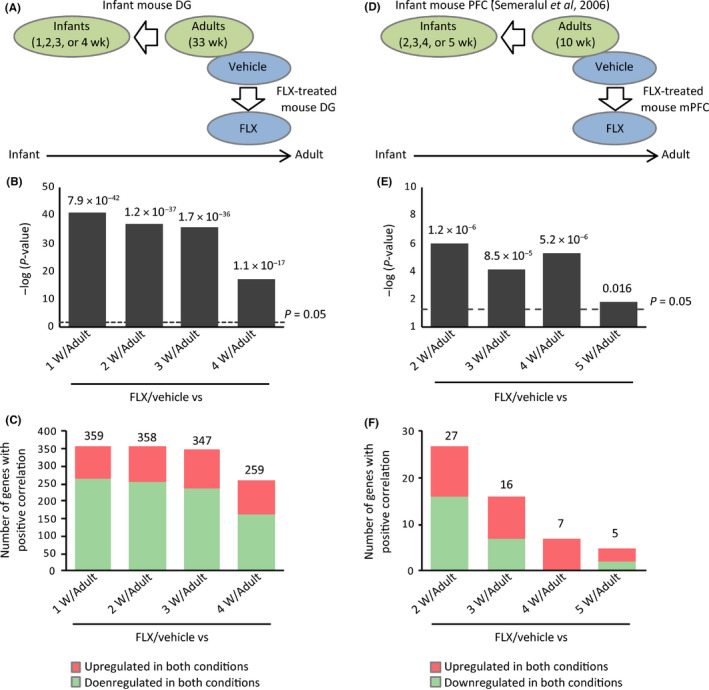
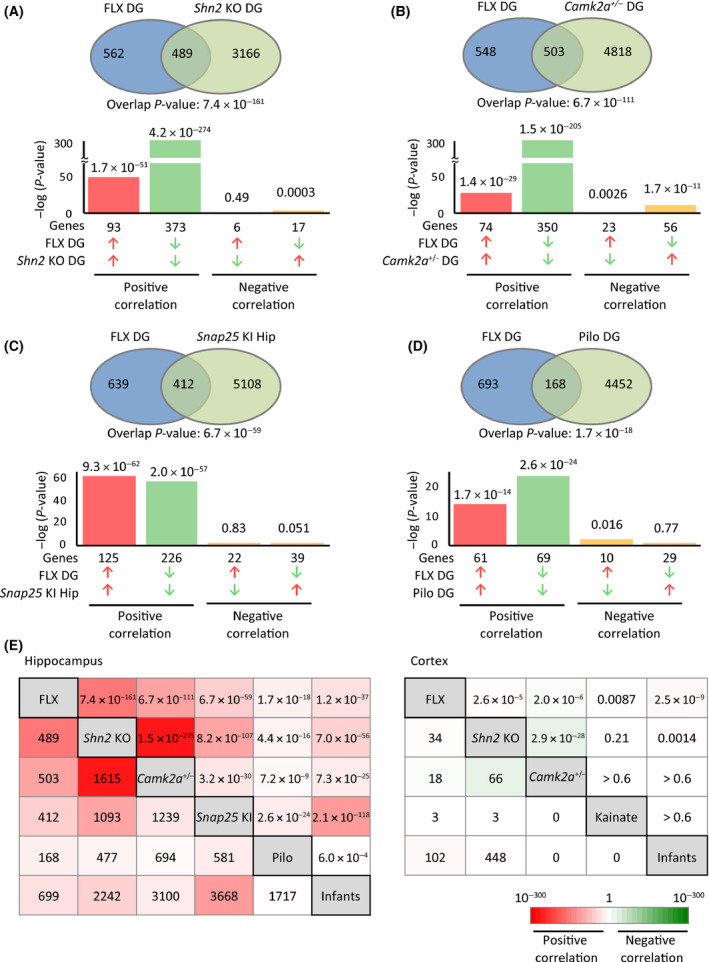
Based on the aforementioned hypothesis, we directly compared transcriptome data between fluoxetine-treated adult mice and those of naive infants in the hippocampus and medial prefrontal cortex (mPFC) to assess similarities and/or differences. We further investigated whether fluoxetine treatment caused dematuration in these brain regions in a hypothesis-free manner using a weighted gene co-expression network analysis (WGCNA).
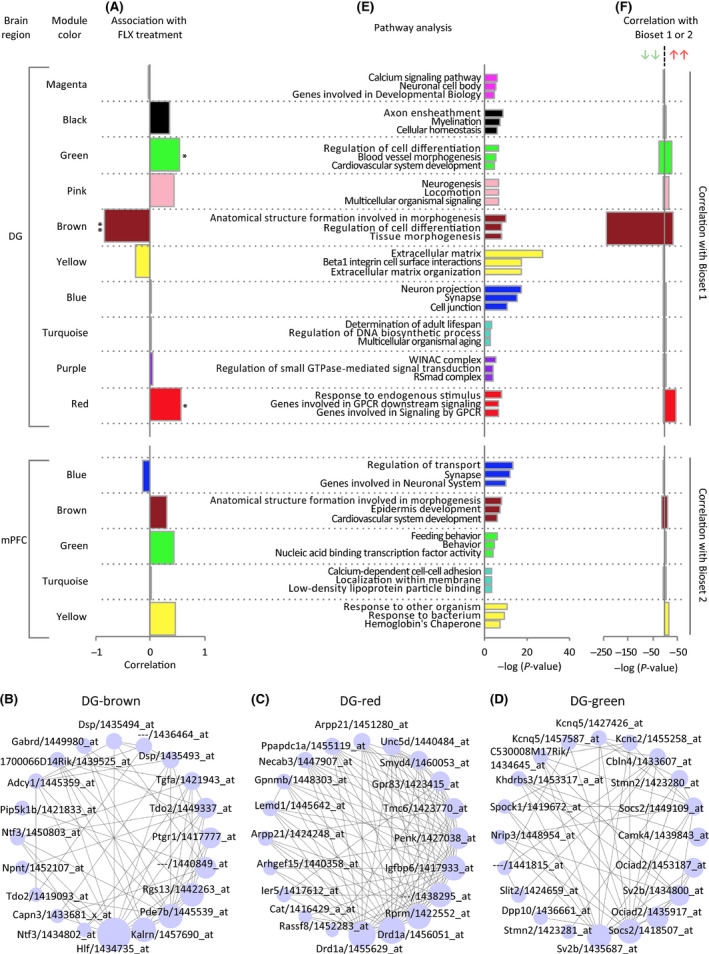
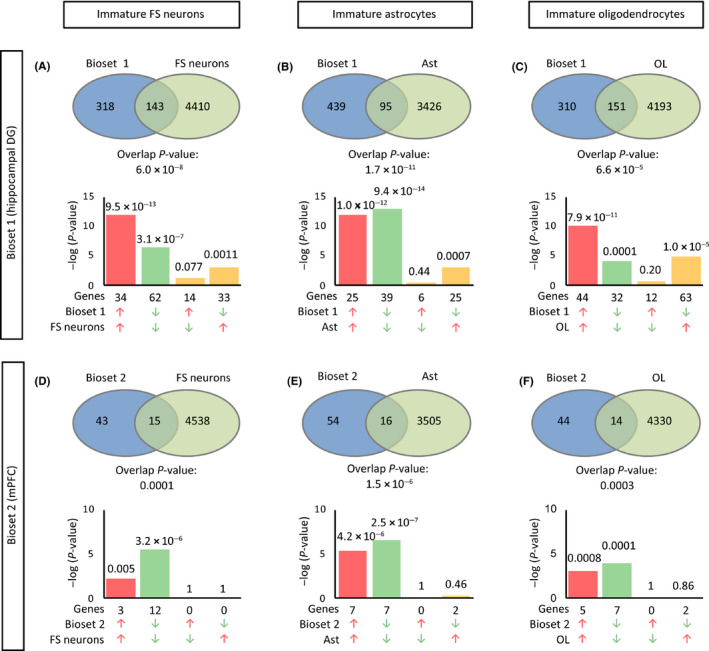
Gene expression patterns in fluoxetine-treated mice resembled those in infants in the mPFC and, to a large extent, in the hippocampus. The gene expression patterns of fluoxetine-treated adult mice were more similar to those of approximately 2-week-old infants than those of older mice. WGCNA confirmed that fluoxetine treatment was associated with maturation abnormalities, particularly in the hippocampus, and highlighted respective co-expression modules for maturity and immaturity marker genes in the hippocampus in response to fluoxetine treatment.
Our results strongly support the hypothesis that chronic fluoxetine treatment induces dematuration in the adult mouse brain from a transcriptomic standpoint. Detection of discrete transcriptomic regulatory networks related to fluoxetine treatment may help to further elucidate the mechanisms of antidepressant action.
氟西汀在大脑中产生抗抑郁作用的分子和细胞机制尚未完全明确。新出现的证据引发了这样一种假说,即慢性氟西汀治疗会导致啮齿动物某些类型的成熟神经元去成熟化。这些研究聚焦于神经元成熟的典型分子和/或电生理标志物的特性。然而,在全基因组基因表达水平上是否存在与去成熟化相关的现象仍不清楚。
基于上述假说,我们直接比较了氟西汀治疗的成年小鼠与未处理的幼鼠在海马体和内侧前额叶皮质(mPFC)中的转录组数据,以评估其异同。我们还使用加权基因共表达网络分析(WGCNA)以无假设的方式进一步研究氟西汀治疗是否会导致这些脑区的去成熟化。
氟西汀治疗小鼠的基因表达模式与mPFC中以及在很大程度上与海马体中幼鼠的基因表达模式相似。氟西汀治疗的成年小鼠的基因表达模式与约2周龄幼鼠的更为相似,而与年龄较大的小鼠不同。WGCNA证实氟西汀治疗与成熟异常有关,特别是在海马体中,并突出了海马体中响应氟西汀治疗的成熟和不成熟标记基因各自的共表达模块。
我们的结果有力地支持了这样一种假说,即从转录组学角度来看,慢性氟西汀治疗会导致成年小鼠大脑去成熟化。检测与氟西汀治疗相关的离散转录组调控网络可能有助于进一步阐明抗抑郁作用的机制。