Signal Transduction laboratory, QIMR Berghofer Medical Research Institute, 300 Herston Road, Herston, Brisbane, QLD, 4006, Australia.
Immunology in Cancer and Infection Laboratory, QIMR Berghofer Medical Research Institute, 300 Herston Road, Herston, Brisbane, QLD, 4006, Australia.
J Exp Clin Cancer Res. 2019 Feb 18;38(1):85. doi: 10.1186/s13046-019-1075-5.
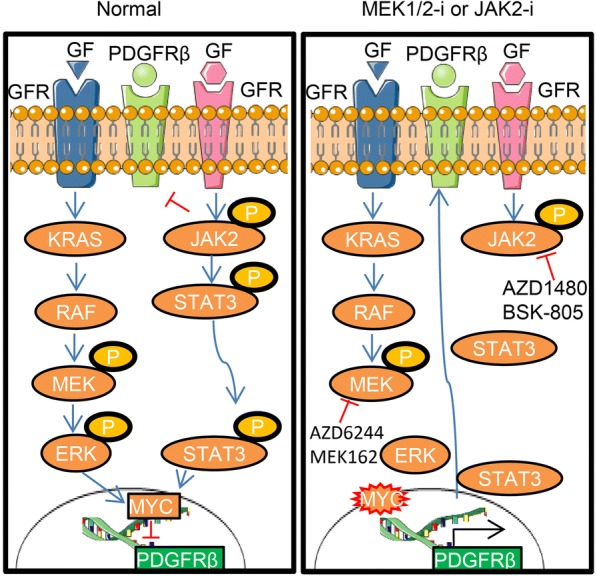
Despite the increasing progress in targeted and immune based-directed therapies for other solid organ malignancies, currently there is no targeted therapy available for TNBCs. A number of mechanisms have been reported both in pre-clinical and clinical settings that involve inherent, acquired and adaptive resistance to small molecule inhibitors. Here, we demonstrated a novel resistance mechanism in TNBC cells mediated by PDGFRβ in response to JAK2 inhibition.
Multiple in vitro (subG1, western blotting, immunofluorescence, RT-PCR, Immunoprecipitation), in vivo and publically available datasets were used.
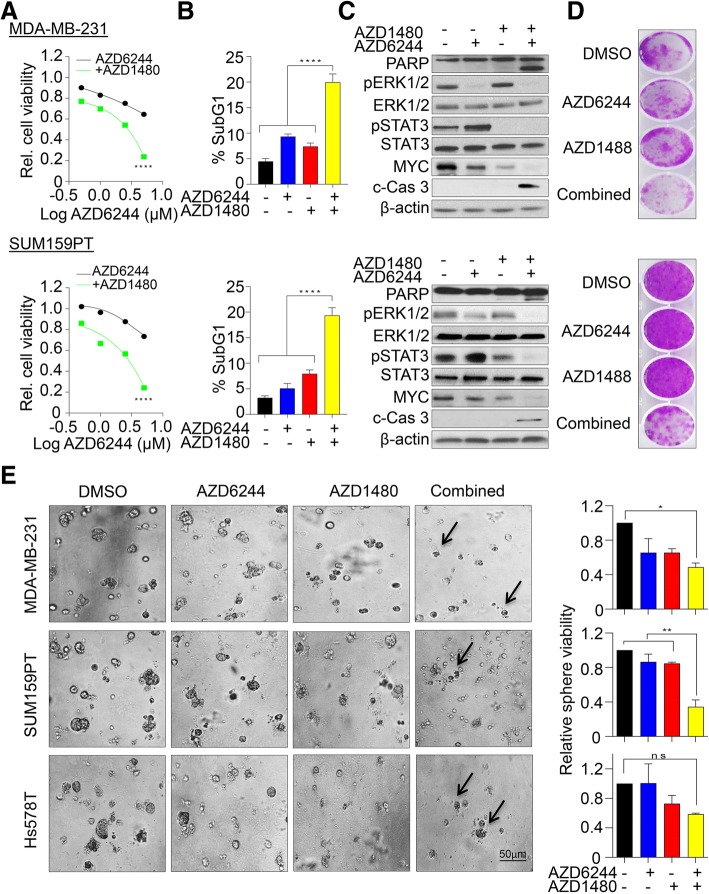
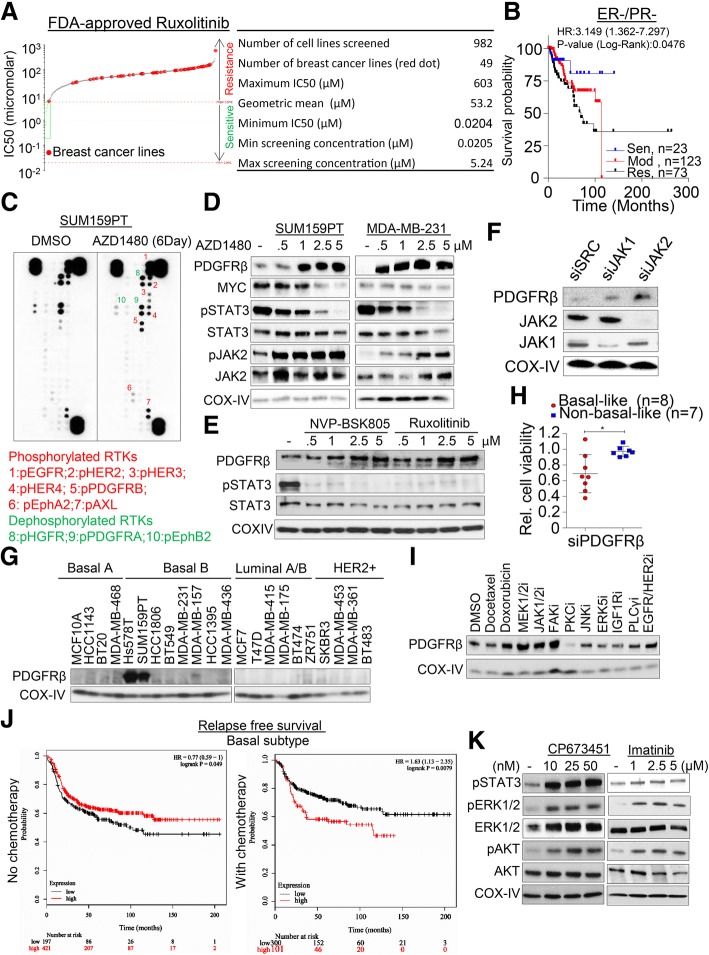
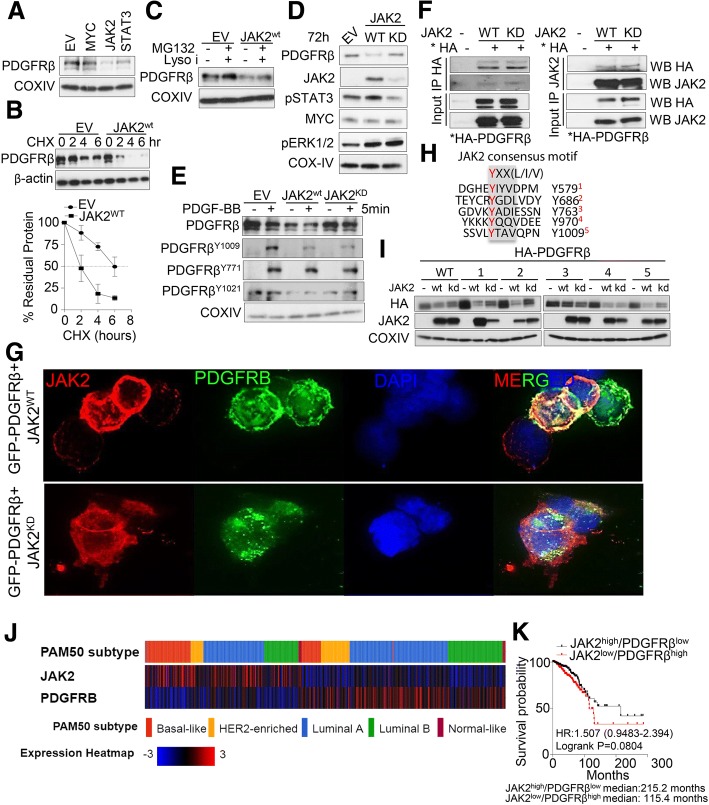
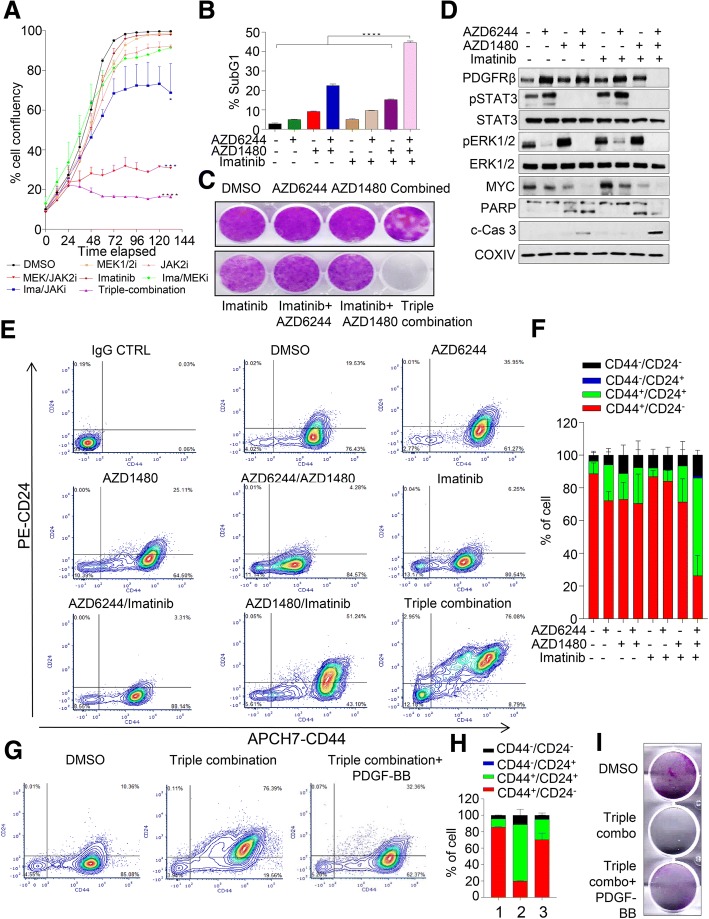
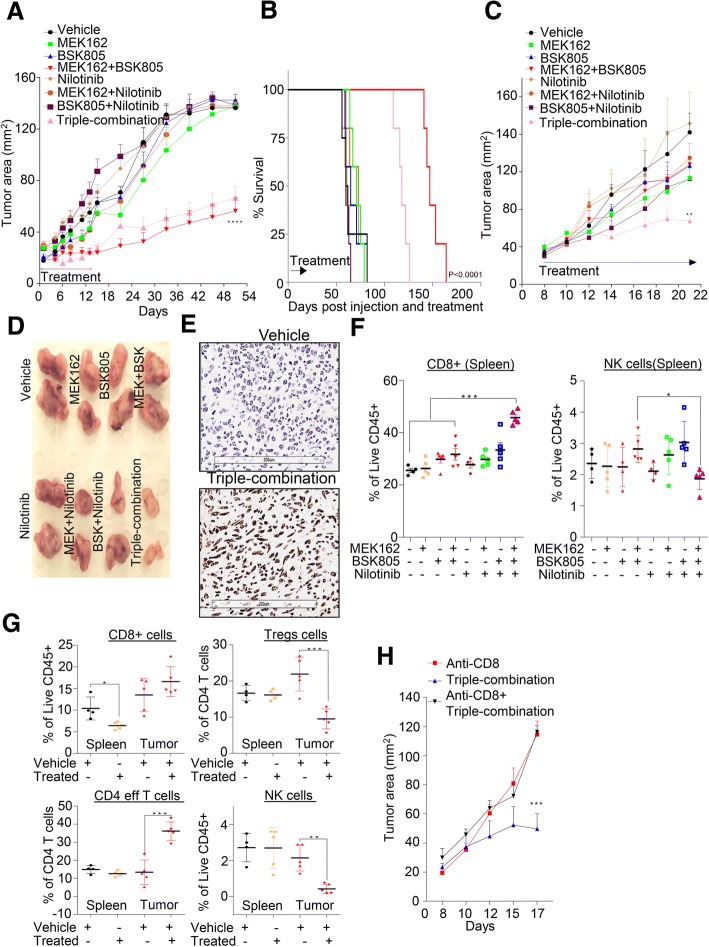
We showed that TNBC cells exposed to MEK1/2-JAK2 inhibitors exhibit resistant colonies in anchorage-independent growth assays. Moreover, cells treated with various small molecule inhibitors including JAK2 promote PDGFRβ upregulation. Using publically available databases, we showed that patients expressing high PDGFRβ or its ligand PDGFB exhibit poor relapse-free survival upon chemotherapeutic treatment. Mechanistically we found that JAK2 expression controls steady state levels of PDGFRβ. Thus, co-blockade of PDGFRβ with JAK2 and MEK1/2 inhibitors completely eradicated resistant colonies in vitro. We found that triple-combined treatment had a significant impact on CD44/CD24 stem-cell-like cells. Likewise, we found a significant tumor growth inhibition in vivo through intratumoral CD8 T cells infiltration in a manner that is reversed by anti-CD8 antibody treatment.
These findings reveal a novel regulatory role of JAK2-mediated PDGFRβ proteolysis and provide an example of a PDGFRβ-mediated resistance mechanism upon specific target inhibition in TNBC.
尽管针对其他实体器官恶性肿瘤的靶向和免疫导向治疗取得了越来越多的进展,但目前尚无针对三阴性乳腺癌(TNBC)的靶向治疗方法。在临床前和临床环境中已经报道了许多涉及小分子抑制剂固有、获得和适应性耐药的机制。在这里,我们证明了 PDGFRβ 在 TNBC 细胞中介导的一种新的耐药机制,以应对 JAK2 抑制。
使用了多种体外(亚 G1、western blot、免疫荧光、RT-PCR、免疫沉淀)、体内和公开可用的数据集。
我们表明,暴露于 MEK1/2-JAK2 抑制剂的 TNBC 细胞在无锚定独立生长测定中表现出耐药集落。此外,用包括 JAK2 在内的各种小分子抑制剂处理的细胞会促进 PDGFRβ 的上调。使用公开可用的数据库,我们表明表达高 PDGFRβ 或其配体 PDGFB 的患者在化疗治疗后表现出较差的无复发生存率。从机制上讲,我们发现 JAK2 表达控制 PDGFRβ 的稳态水平。因此,PDGFRβ 与 JAK2 和 MEK1/2 抑制剂的联合阻断完全消除了体外耐药集落。我们发现三联治疗对 CD44/CD24 干细胞样细胞有显著影响。同样,我们发现通过肿瘤内 CD8 T 细胞浸润在体内显著抑制肿瘤生长,这种作用可通过抗 CD8 抗体治疗逆转。
这些发现揭示了 JAK2 介导的 PDGFRβ 蛋白水解的新调节作用,并提供了一个在 TNBC 中针对特定靶标抑制时 PDGFRβ 介导的耐药机制的例子。