Peces Ramón, Mena Rocío, Peces Carlos, Santos-Simarro Fernando, Fernández Luis, Afonso Sara, Lapunzina Pablo, Selgas Rafael, Nevado Julián
Nephrology Department, La Paz University Hospital, IdiPAZ, Autonomous University, Madrid, Spain.
La Paz University Hospital, Medical and Molecular Genetics Institute (INGEMM), IdiPAZ, Madrid, Spain.
Mol Genet Genomic Med. 2019 Apr;7(4):e00568. doi: 10.1002/mgg3.568. Epub 2019 Feb 19.
Congenital nephrogenic diabetes insipidus (NDI) is a rare condition characterized by severe polyuria, due to the inability of the kidneys to concentrate urine in response to arginine vasopressin (AVP). In the majority of the cases, the disease shows an X-linked inherited pattern, although an autosomal recessive inheritance was also observed.
We report a patient with a severe NDI diagnosed during the neonatal period. Because the patient was female without a family history of congenital NDI, her disease was thought to exhibit an autosomal recessive form.
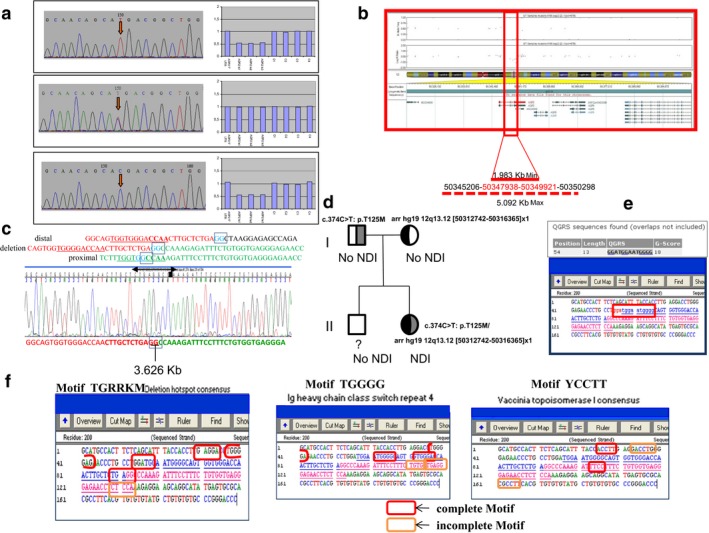
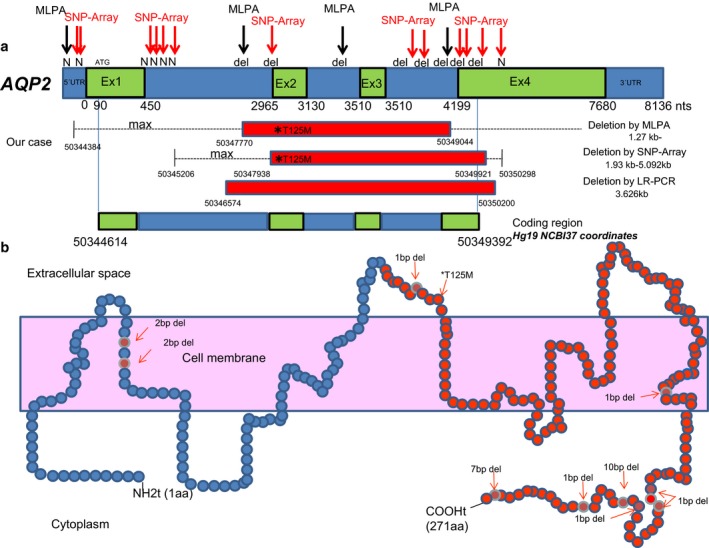
A full mutation analysis of AVP receptor 2 (AVPR2; MIM#300538) gene showed no mutations. However, direct Sanger sequencing of the aquaporin 2 (AQP2) revealed an apparently homozygous mutation at nucleotide position NM_000486.5:c.374C>T (p.Thr125Met) in exon 2. Further customized multiplex ligation-dependent probe amplification (MLPA), single-nucleotide polymorphism (SNP) array analysis, and long-range polymerase chain reaction (PCR) followed by Sanger sequencing showed a heterozygous exonic deletion comprising exons 2, 3, and partially 4 of AQP2.
This is the first case of a compound heterozygote patient with a missense mutation involving NM_000486.5:exon2:c.374C>T (p.Thr125Met) and a gross deletion of at least exons 2, 3, and partially 4 on the AQP2 to present with a severe NDI phenotype.
先天性肾性尿崩症(NDI)是一种罕见病症,其特征为严重多尿,这是由于肾脏无法对精氨酸加压素(AVP)作出反应而浓缩尿液所致。在大多数病例中,该疾病呈X连锁遗传模式,不过也观察到常染色体隐性遗传情况。
我们报告了一名在新生儿期被诊断出患有严重NDI的患者。由于该患者为女性且无先天性NDI家族史,其疾病被认为呈现常染色体隐性形式。
对AVP受体2(AVPR2;MIM#300538)基因进行的全面突变分析未发现突变。然而,对水通道蛋白2(AQP2)进行直接桑格测序显示,在第2外显子的核苷酸位置NM_000486.5:c.374C>T(p.Thr125Met)处存在一个明显的纯合突变。进一步进行定制的多重连接依赖探针扩增(MLPA)、单核苷酸多态性(SNP)阵列分析以及长距离聚合酶链反应(PCR),随后进行桑格测序,结果显示存在一个杂合性外显子缺失,包括AQP2的第2、3外显子以及部分第4外显子。
这是首例患有错义突变(涉及NM_000486.5:外显子2:c.374C>T(p.Thr125Met))和AQP2至少第2、3外显子以及部分第4外显子大片段缺失的复合杂合子患者,该患者表现出严重的NDI表型。