Groupe d'Étude des Protéines Membranaires (GÉPROM), Département de Physiologie, Université de Montréal, Montréal, Québec, Canada ; Centre de Recherche, Hôpital du Sacré-Cœur de Montréal, Montréal, Québec, Canada.
Groupe d'Étude des Protéines Membranaires (GÉPROM), Département de Physiologie, Université de Montréal, Montréal, Québec, Canada.
Clin Kidney J. 2012 Jun;5(3):195-202. doi: 10.1093/ckj/sfs029. Epub 2012 Mar 28.
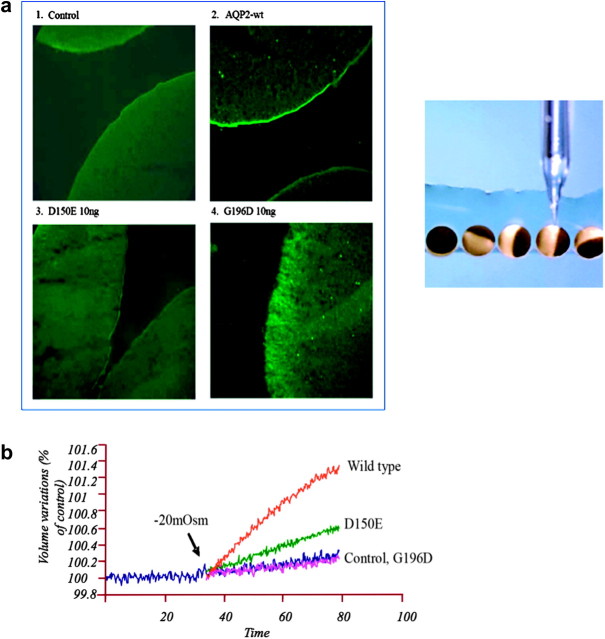
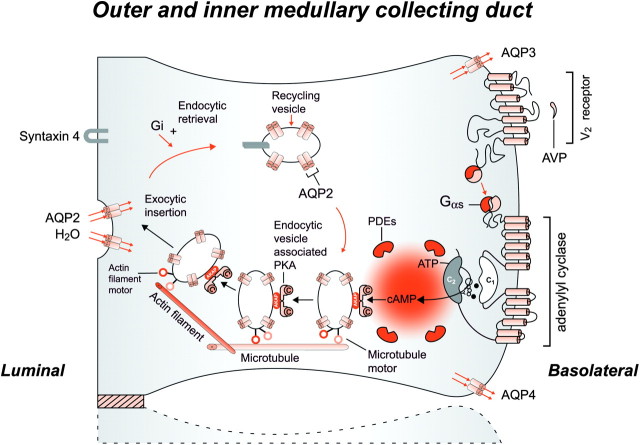
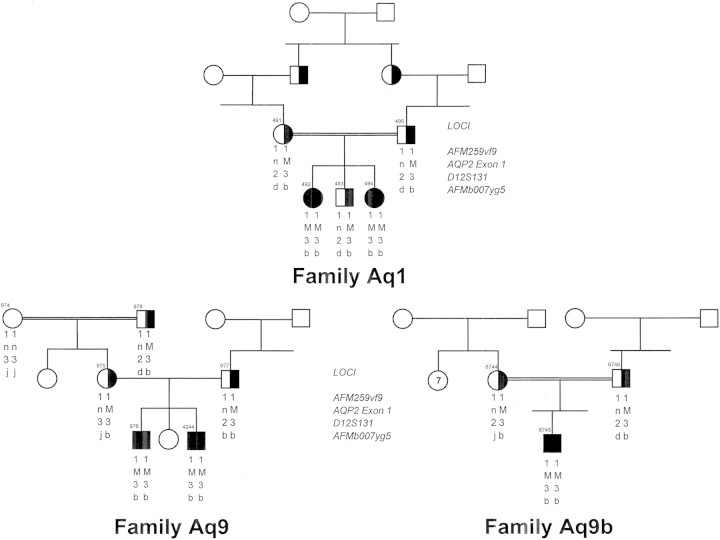
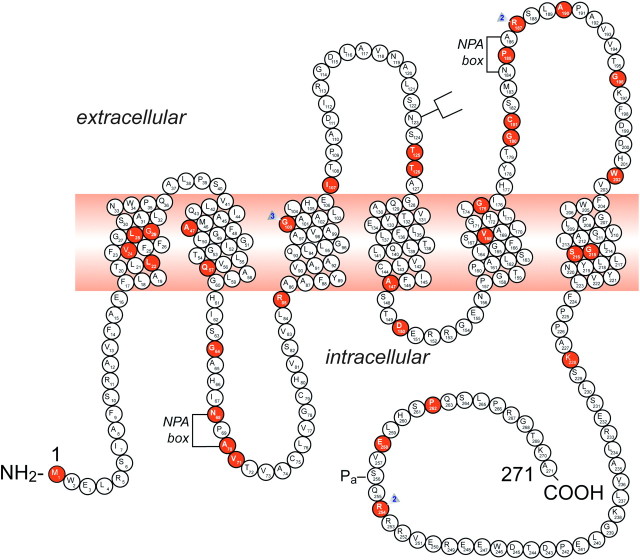
It is clinically useful to distinguish between two types of hereditary nephrogenic diabetes insipidus (NDI): a 'pure' type characterized by loss of water only and a complex type characterized by loss of water and ions. Patients with congenital NDI bearing mutations in the vasopressin 2 receptor gene, AVPR2, or in the aquaporin-2 gene, AQP2, have a pure NDI phenotype with loss of water but normal conservation of sodium, potassium, chloride and calcium. Patients with hereditary hypokalemic salt-losing tubulopathies have a complex phenotype with loss of water and ions. They have polyhydramnios, hypercalciuria and hypo- or isosthenuria and were found to bear KCNJ1 (ROMK) and SLC12A1 (NKCC2) mutations. Patients with polyhydramnios, profound polyuria, hyponatremia, hypochloremia, metabolic alkalosis and sensorineural deafness were found to bear BSND mutations. These clinical phenotypes demonstrate the critical importance of the proteins ROMK, NKCC2 and Barttin to transfer NaCl in the medullary interstitium and thereby to generate, together with urea, a hypertonic milieu. This editorial describes two new developments: (i) the genomic information provided by the sequencing of the AQP2 gene is key to the routine care of these patients, and, as in other genetic diseases, reduces health costs and provides psychological benefits to patients and families and (ii) the expression of AQP2 mutants in Xenopus oocytes and in polarized renal tubular cells recapitulates the clinical phenotypes and reveals a continuum from severe loss of function with urinary osmolalities <150 mOsm/kg H2O to milder defects with urine osmolalities >200 mOsm/kg H2O.
临床上有用的区分两种类型的遗传性肾性尿崩症(NDI):一种“纯”型,其特征是仅丧失水,另一种是复杂型,其特征是丧失水和离子。患有先天性 NDI 的患者,其突变位于血管加压素 2 受体基因(AVPR2)或水通道蛋白 2 基因(AQP2),具有纯 NDI 表型,丧失水但钠、钾、氯和钙正常保留。遗传性低钾性失盐性肾小管病患者具有复杂表型,丧失水和离子。他们有羊水过多、高钙尿症和低尿或等尿,并且发现携带 KCNJ1(ROMK)和 SLC12A1(NKCC2)突变。羊水过多、严重多尿、低钠血症、低氯血症、代谢性碱中毒和感觉神经性耳聋的患者发现携带 BSND 突变。这些临床表型表明 ROMK、NKCC2 和 Barttin 蛋白对转移在髓质间质中的 NaCl 的重要性,从而与尿素一起产生高渗环境。这篇社论描述了两个新的发展:(i)AQP2 基因测序提供的基因组信息对这些患者的常规护理至关重要,并且,与其他遗传疾病一样,降低了医疗费用,并为患者和家庭提供了心理益处;(ii)在非洲爪蟾卵母细胞和极化肾小管细胞中表达 AQP2 突变体再现了临床表型,并揭示了从严重功能丧失(尿渗透压 <150 mOsm/kg H2O)到更轻度缺陷(尿渗透压 >200 mOsm/kg H2O)的连续谱。