Department of Physiological Chemistry, University of Veterinary Medicine Hannover, Bünteweg 17, 30559 Hannover, Germany.
Laboratory of Functional Physiology and Valorization of Bioresources⁻Higher Institute of Biotechnology of Béja, University of Jendouba, Jendouba 8189, Tunisia.
Nutrients. 2019 Feb 22;11(2):461. doi: 10.3390/nu11020461.
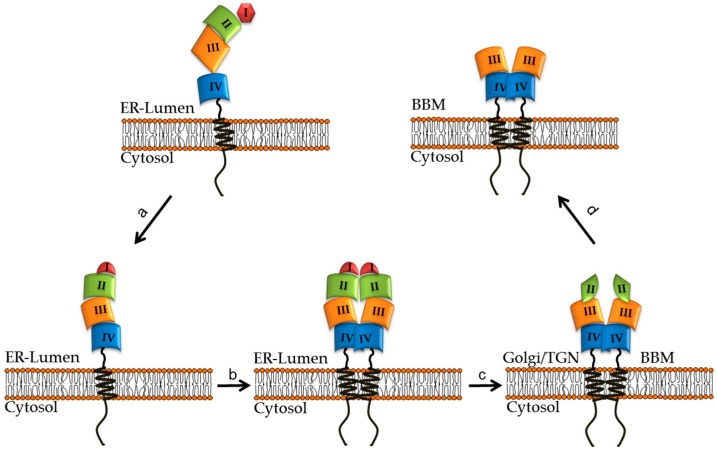
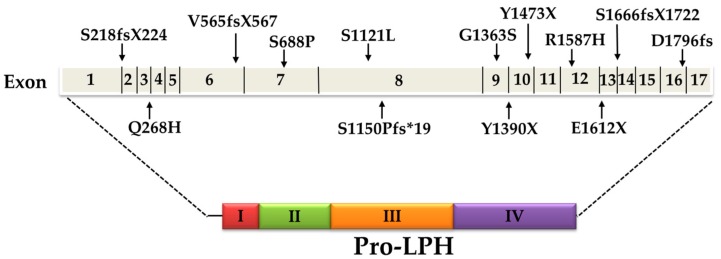
Congenital lactase deficiency (CLD) is a severe autosomal recessive genetic disorder that affects the functional capacity of the intestinal protein lactase-phlorizin hydrolase (LPH). This disorder is diagnosed already during the first few days of the newborn's life due to the inability to digest lactose, the main carbohydrate in mammalian milk. The symptoms are similar to those in other carbohydrate malabsorption disorders, such as congenital sucrase-isomaltase deficiency, and include severe osmotic watery diarrhea. CLD is associated with mutations in the translated region of the LPH gene that elicit loss-of-function of LPH. The mutations occur in a homozygote or compound heterozygote pattern of inheritance and comprise missense mutations as well as mutations that lead to complete or partial truncations of crucial domains in LPH, such as those linked to the folding and transport-competence of LPH and to the catalytic domains. Nevertheless, the identification of the mutations in CLD is not paralleled by detailed genotype/protein phenotype analyses that would help unravel potential pathomechanisms underlying this severe disease. Here, we review the current knowledge of CLD mutations and discuss their potential impact on the structural and biosynthetic features of LPH. We also address the question of whether heterozygote carriers can be symptomatic for CLD and whether genetic testing is needed in view of the severity of the disease.
先天性乳糖酶缺乏症(CLD)是一种严重的常染色体隐性遗传疾病,影响肠道蛋白乳糖酶-漆酶水解酶(LPH)的功能能力。由于不能消化乳糖,这种疾病在新生儿生命的头几天就已经被诊断出来,乳糖是哺乳动物奶中的主要碳水化合物。其症状与其他碳水化合物吸收不良疾病相似,如先天性蔗糖酶-异麦芽糖酶缺乏症,包括严重的渗透水样腹泻。CLD 与 LPH 基因翻译区域的突变有关,这些突变导致 LPH 失去功能。突变以纯合子或复合杂合子的遗传模式发生,包括错义突变以及导致 LPH 中关键结构域完全或部分截断的突变,例如与 LPH 的折叠和运输能力以及催化结构域相关的突变。然而,CLD 突变的鉴定并没有伴随着详细的基因型/蛋白表型分析,这有助于阐明这种严重疾病潜在的发病机制。在这里,我们回顾了 CLD 突变的现有知识,并讨论了它们对 LPH 的结构和生物合成特征的潜在影响。我们还探讨了杂合子携带者是否会出现 CLD 症状的问题,以及鉴于疾病的严重程度,是否需要进行基因检测。