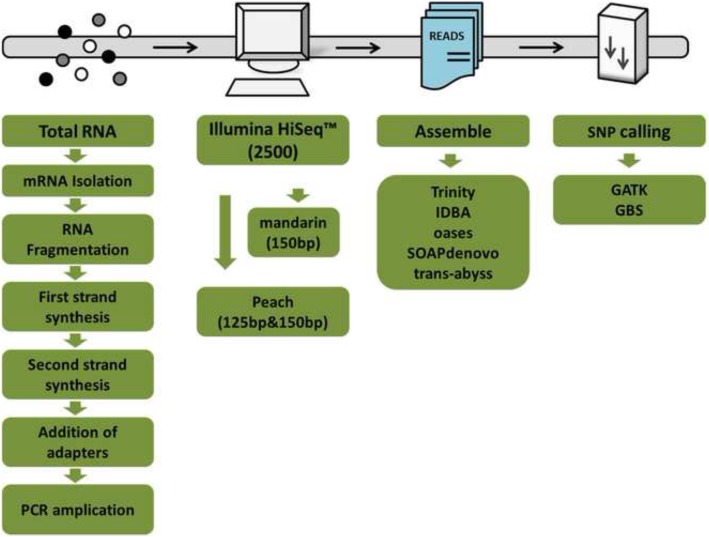
Zhejiang Provincial Key Laboratory of Horticultural Plant Integrative Biology, Zhejiang University, Zijingang Campus, Hangzhou, China.
BMC Genomics. 2019 Feb 27;20(1):160. doi: 10.1186/s12864-019-5533-4.
Single nucleotide polymorphisms (SNP) have been applied as important molecular markers in genetics and breeding studies. The rapid advance of next generation sequencing (NGS) provides a high-throughput means of SNP discovery. However, SNP development is limited by the availability of reliable SNP discovery methods. Especially, the optimum assembler and SNP caller for accurate SNP prediction from next generation sequencing data are not known.
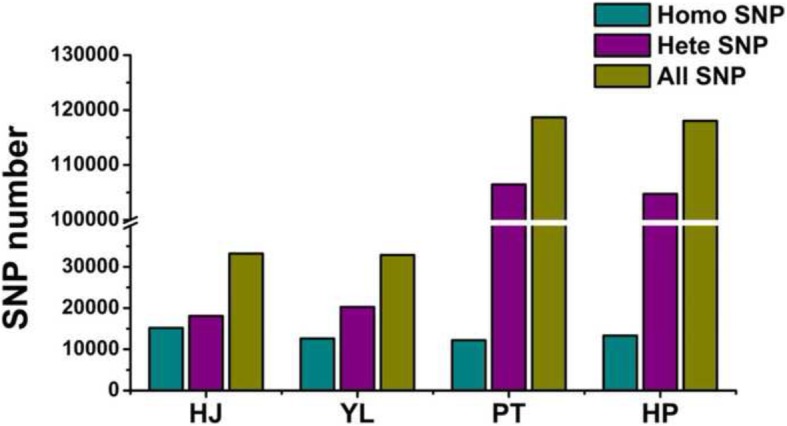
Herein we performed SNP prediction based on RNA-seq data of peach and mandarin peel tissue under a comprehensive comparison of two paired-end read lengths (125 bp and 150 bp), five assemblers (Trinity, IDBA, oases, SOAPdenovo, Trans-abyss) and two SNP callers (GATK and GBS). The predicted SNPs were compared with the authentic SNPs identified via PCR amplification followed by gene cloning and sequencing procedures. A total of 40 and 240 authentic SNPs were presented in five anthocyanin biosynthesis related genes in peach and in nine carotenogenic genes in mandarin. Putative SNPs predicted from the same RNA-seq data with different strategies led to quite divergent results. The rate of false positive SNPs was significantly lower when the paired-end read length was 150 bp compared with 125 bp. Trinity was superior to the other four assemblers and GATK was substantially superior to GBS due to a low rate of missing authentic SNPs. The combination of assembler Trinity, SNP caller GATK, and the paired-end read length 150 bp had the best performance in SNP discovery with 100% accuracy both in peach and in mandarin cases. This strategy was applied to the characterization of SNPs in peach and mandarin transcriptomes.
Through comparison of authentic SNPs obtained by PCR cloning strategy and putative SNPs predicted from different combinations of five assemblers, two SNP callers, and two paired-end read lengths, we provided a reliable and efficient strategy, Trinity-GATK with 150 bp paired-end read length, for SNP discovery from RNA-seq data. This strategy discovered SNP at 100% accuracy in peach and mandarin cases and might be applicable to a wide range of plants and other organisms.
单核苷酸多态性(SNP)已被用作遗传学和育种研究中的重要分子标记。下一代测序(NGS)的快速发展提供了一种高通量的 SNP 发现方法。然而,SNP 的开发受到可靠的 SNP 发现方法的限制。特别是,用于从下一代测序数据中准确预测 SNP 的最佳组装程序和 SNP 调用程序尚不清楚。
在此,我们通过比较两种不同的双端读长(125bp 和 150bp)、五种组装程序(Trinity、IDBA、oases、SOAPdenovo、Trans-abyss)和两种 SNP 调用程序(GATK 和 GBS),在桃和柑橘皮组织的 RNA-seq 数据上进行了 SNP 预测。将预测的 SNP 与通过 PCR 扩增、基因克隆和测序程序鉴定的真实 SNP 进行比较。在五个花青素生物合成相关基因和九个类胡萝卜素生物合成基因中,共鉴定出 40 个和 240 个真实 SNP。使用不同策略从相同的 RNA-seq 数据中预测的假定 SNP 导致了截然不同的结果。与 125bp 相比,150bp 双端读长的假阳性 SNP 率显著降低。Trinity 优于其他四个组装程序,GATK 由于真实 SNP 缺失率低,明显优于 GBS。组装程序 Trinity、SNP 调用程序 GATK 和 150bp 双端读长的组合在 SNP 发现方面表现最佳,在桃和柑橘的情况下准确率均为 100%。该策略应用于桃和柑橘转录组中 SNP 的表征。
通过比较通过 PCR 克隆策略获得的真实 SNP 和从五种组装程序、两种 SNP 调用程序和两种双端读长的不同组合中预测的假定 SNP,我们提供了一种可靠且高效的策略,即使用 Trinity-GATK 和 150bp 双端读长进行 SNP 发现。该策略在桃和柑橘的情况下 SNP 发现准确率为 100%,可能适用于广泛的植物和其他生物。