Department of Mathematics and Statistics and Organismal and Evolutionary Biology Research Programme, University of Helsinki, Helsinki, Finland.
CSIRO Agriculture & Food, GPO Box 1600, Canberra, ACT 2601, Australia.
Bioinformatics. 2019 Oct 1;35(19):3684-3692. doi: 10.1093/bioinformatics/btz164.
Recent advances in high dimensional phenotyping bring time as an extra dimension into the phenotypes. This promotes the quantitative trait locus (QTL) studies of function-valued traits such as those related to growth and development. Existing approaches for analyzing functional traits utilize either parametric methods or semi-parametric approaches based on splines and wavelets. However, very limited choices of software tools are currently available for practical implementation of functional QTL mapping and variable selection.
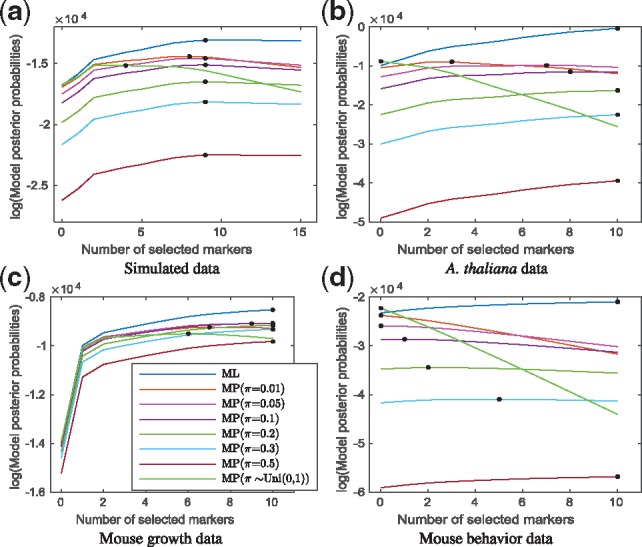
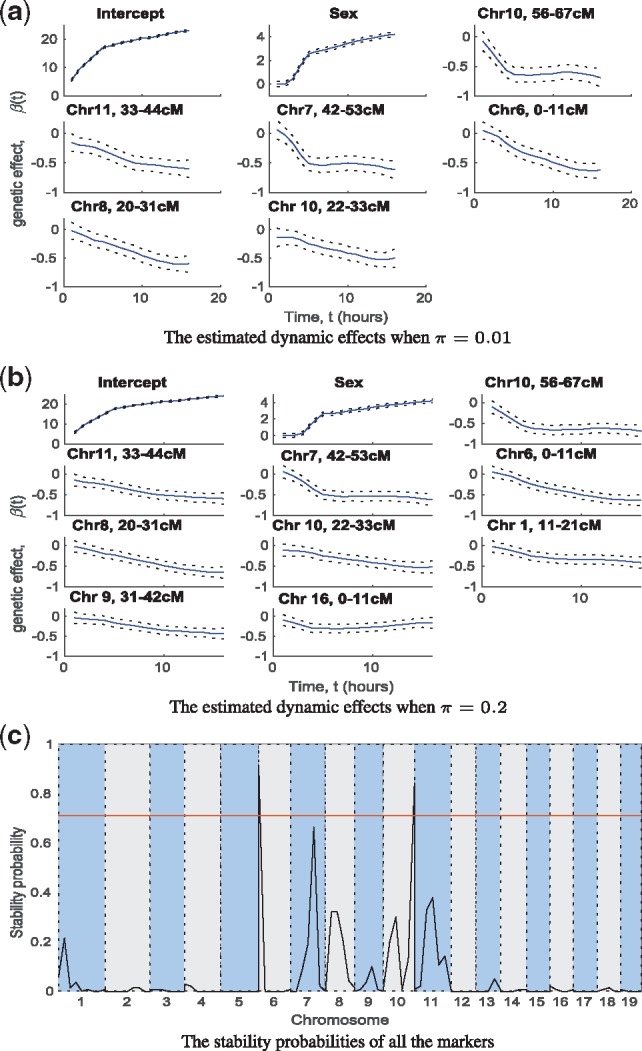
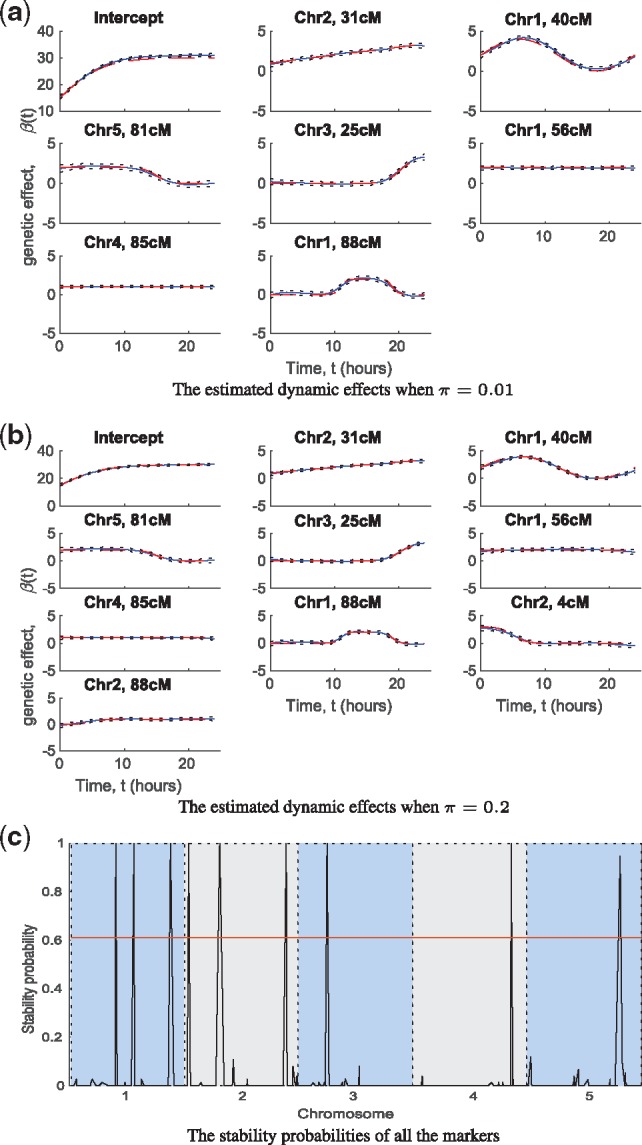
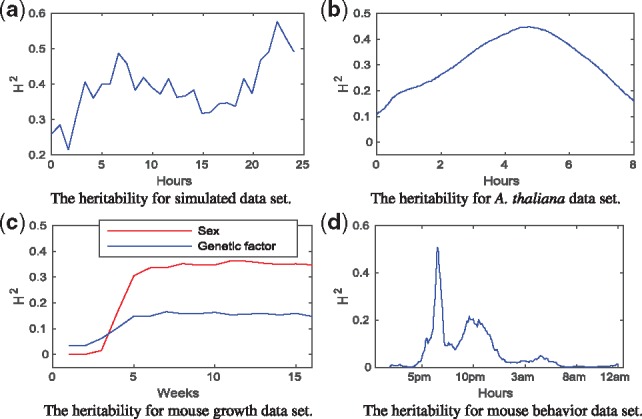
We propose a Bayesian Gaussian process (GP) approach for functional QTL mapping. We use GPs to model the continuously varying coefficients which describe how the effects of molecular markers on the quantitative trait are changing over time. We use an efficient gradient based algorithm to estimate the tuning parameters of GPs. Notably, the GP approach is directly applicable to the incomplete datasets having even larger than 50% missing data rate (among phenotypes). We further develop a stepwise algorithm to search through the model space in terms of genetic variants, and use a minimal increase of Bayesian posterior probability as a stopping rule to focus on only a small set of putative QTL. We also discuss the connection between GP and penalized B-splines and wavelets. On two simulated and three real datasets, our GP approach demonstrates great flexibility for modeling different types of phenotypic trajectories with low computational cost. The proposed model selection approach finds the most likely QTL reliably in tested datasets.
Software and simulated data are available as a MATLAB package 'GPQTLmapping', and they can be downloaded from GitHub (https://github.com/jpvanhat/GPQTLmapping). Real datasets used in case studies are publicly available at QTL Archive.
Supplementary data are available at Bioinformatics online.
高维表型分析的最新进展将时间作为一个额外的维度引入到表型中。这促进了功能值性状(如与生长和发育相关的性状)的数量性状基因座(QTL)研究。现有的分析功能性状的方法要么利用参数方法,要么利用基于样条和小波的半参数方法。然而,目前用于功能 QTL 映射和变量选择的实用实现的软件工具选择非常有限。
我们提出了一种用于功能 QTL 映射的贝叶斯高斯过程(GP)方法。我们使用 GPs 来建模描述分子标记对定量性状的影响随时间变化的连续变化系数。我们使用有效的基于梯度的算法来估计 GPs 的调整参数。值得注意的是,GP 方法可直接应用于具有超过 50%缺失数据率(在表型中)的不完整数据集。我们进一步开发了一种逐步算法,根据遗传变异搜索模型空间,并使用贝叶斯后验概率的最小增加作为停止规则,只关注一小部分假定的 QTL。我们还讨论了 GP 与惩罚 B 样条和小波之间的关系。在两个模拟数据集和三个真实数据集上,我们的 GP 方法以低计算成本展示了对不同类型的表型轨迹进行建模的极大灵活性。所提出的模型选择方法在测试数据集上可靠地找到了最可能的 QTL。
软件和模拟数据作为 MATLAB 包“GPQTLmapping”提供,可从 GitHub(https://github.com/jpvanhat/GPQTLmapping)下载。案例研究中使用的真实数据集可在 QTL 档案中公开获得。
补充数据可在生物信息学在线获得。