Center for Bioinformatics, Saarland University, Saarland Informatics Campus E2.1, 66123 Saarbrücken, Germany.
Max Planck Institute for Informatics, Saarland Informatics Campus E1.4, 66123 Saarbrücken, Germany.
Bioinformatics. 2019 Oct 1;35(19):3599-3607. doi: 10.1093/bioinformatics/btz162.
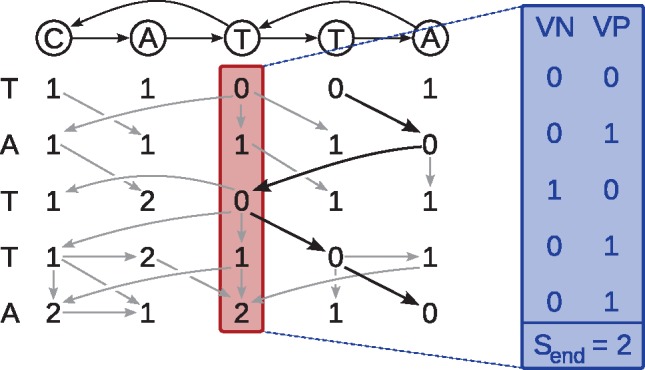
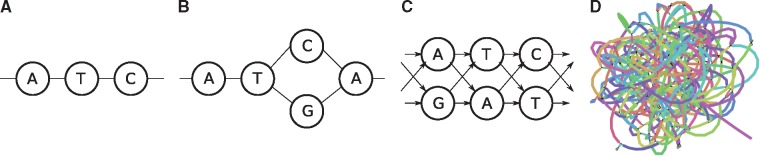
Graphs are commonly used to represent sets of sequences. Either edges or nodes can be labeled by sequences, so that each path in the graph spells a concatenated sequence. Examples include graphs to represent genome assemblies, such as string graphs and de Bruijn graphs, and graphs to represent a pan-genome and hence the genetic variation present in a population. Being able to align sequencing reads to such graphs is a key step for many analyses and its applications include genome assembly, read error correction and variant calling with respect to a variation graph.
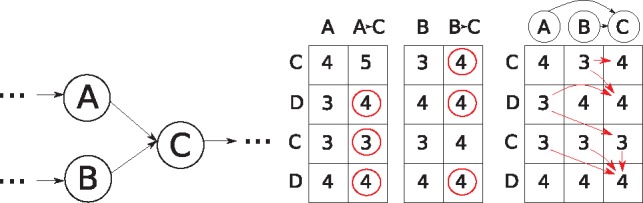
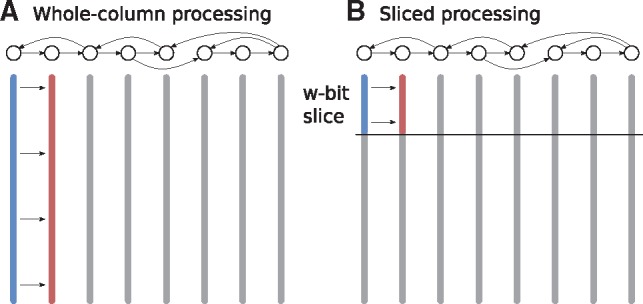
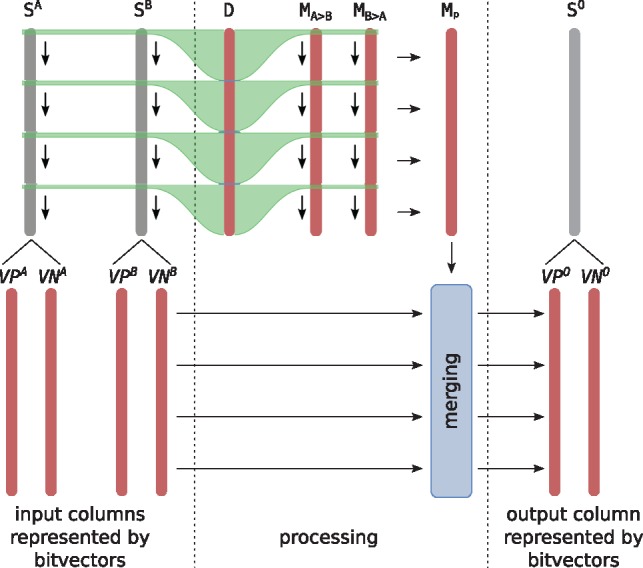
We generalize two linear sequence-to-sequence algorithms to graphs: the Shift-And algorithm for exact matching and Myers' bitvector algorithm for semi-global alignment. These linear algorithms are both based on processing w sequence characters with a constant number of operations, where w is the word size of the machine (commonly 64), and achieve a speedup of up to w over naive algorithms. For a graph with |V| nodes and |E| edges and a sequence of length m, our bitvector-based graph alignment algorithm reaches a worst case runtime of O(|V|+⌈mw⌉|E| log w) for acyclic graphs and O(|V|+m|E| log w) for arbitrary cyclic graphs. We apply it to five different types of graphs and observe a speedup between 3-fold and 20-fold compared with a previous (asymptotically optimal) alignment algorithm.
https://github.com/maickrau/GraphAligner.
Supplementary data are available at Bioinformatics online.
图通常用于表示序列集。边或节点都可以由序列标记,因此图中的每条路径都拼写一个连接的序列。例如,包括表示基因组组装的图,如字符串图和 de Bruijn 图,以及表示泛基因组的图,因此表示种群中存在的遗传变异。能够将测序读取与这些图对齐是许多分析的关键步骤,其应用包括基因组组装、读取错误校正和针对变异图的变异调用。
我们将两种线性序列到序列算法推广到图中:用于精确匹配的 Shift-And 算法和用于半全局对齐的 Myers 位向量算法。这两种线性算法都是基于使用固定数量的操作处理 w 个序列字符,其中 w 是机器的字大小(通常为 64),并实现高达 w 的速度提升超过了原始算法。对于具有 |V| 个节点和 |E| 条边且长度为 m 的序列,我们基于位向量的图对齐算法在非循环图中的最坏情况运行时间为 O(|V|+⌈mw⌉|E|logw),在任意循环图中的运行时间为 O(|V|+m|E|logw)。我们将其应用于五种不同类型的图,并观察到与以前的(渐近最优)对齐算法相比,速度提高了 3 倍到 20 倍。
https://github.com/maickrau/GraphAligner。
补充数据可在 Bioinformatics 在线获得。