Fu Xiangzheng, Zhu Wen, Cai Lijun, Liao Bo, Peng Lihong, Chen Yifan, Yang Jialiang
College of Information Science and Engineering, Hunan University, Changsha, China.
School of Mathematics and Statistics, Hainan Normal University, Haikou, China.
Front Genet. 2019 Feb 25;10:119. doi: 10.3389/fgene.2019.00119. eCollection 2019.
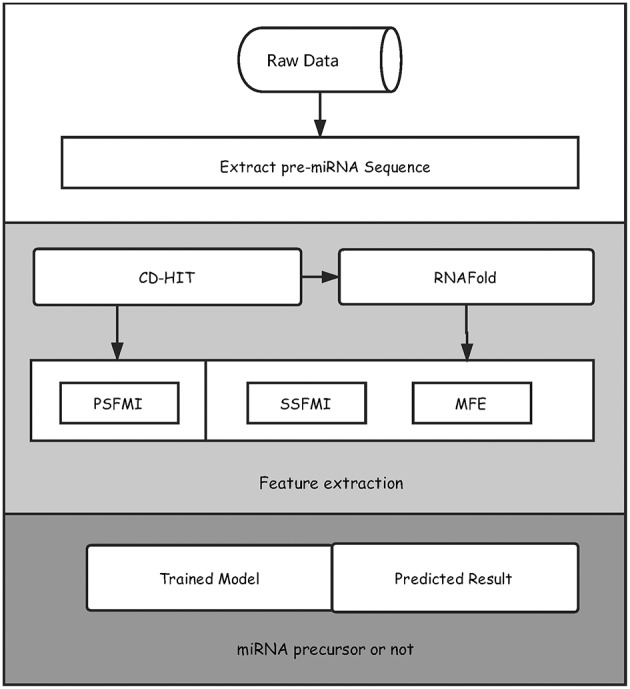
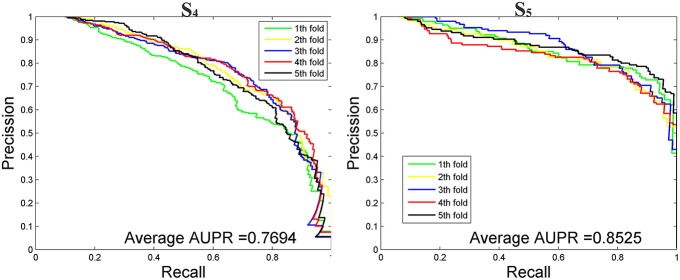
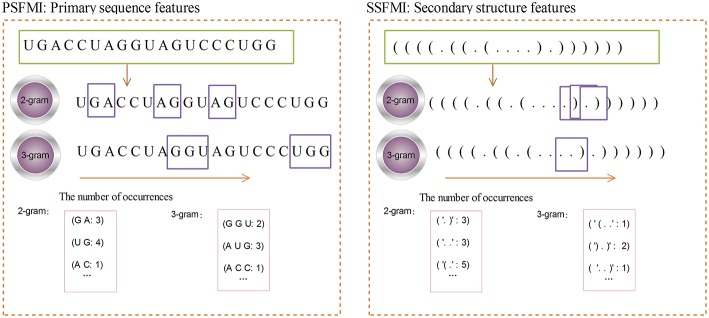
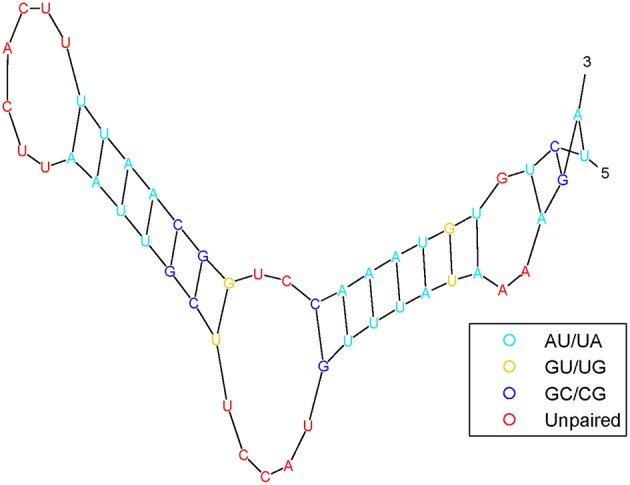
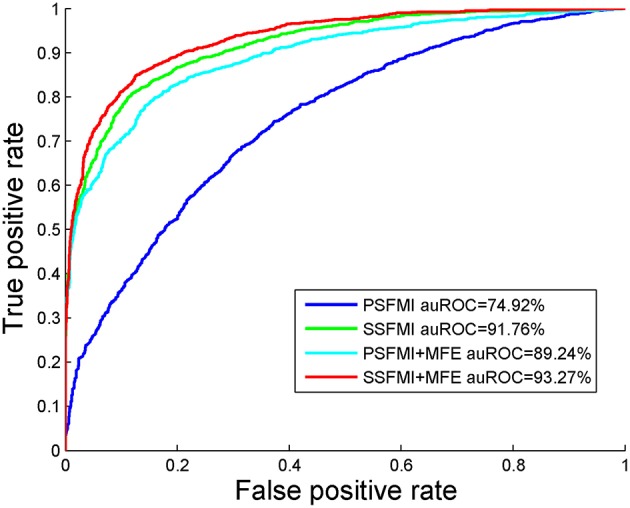
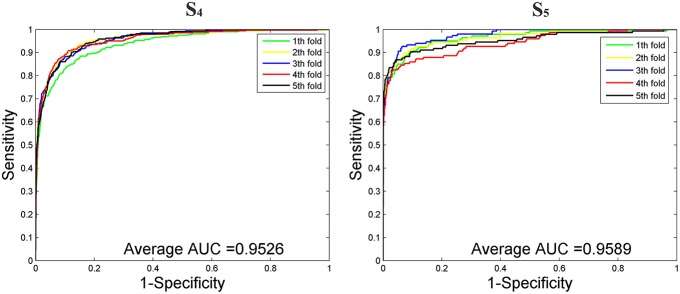
Playing critical roles as post-transcriptional regulators, microRNAs (miRNAs) are a family of short non-coding RNAs that are derived from longer transcripts called precursor miRNAs (pre-miRNAs). Experimental methods to identify pre-miRNAs are expensive and time-consuming, which presents the need for computational alternatives. In recent years, the accuracy of computational methods to predict pre-miRNAs has been increasing significantly. However, there are still several drawbacks. First, these methods usually only consider base frequencies or sequence information while ignoring the information between bases. Second, feature extraction methods based on secondary structures usually only consider the global characteristics while ignoring the mutual influence of the local structures. Third, methods integrating high-dimensional feature information is computationally inefficient. In this study, we have proposed a novel mutual information-based feature representation algorithm for pre-miRNA sequences and secondary structures, which is capable of catching the interactions between sequence bases and local features of the RNA secondary structure. In addition, the feature space is smaller than that of most popular methods, which makes our method computationally more efficient than the competitors. Finally, we applied these features to train a support vector machine model to predict pre-miRNAs and compared the results with other popular predictors. As a result, our method outperforms others based on both 5-fold cross-validation and the Jackknife test.
微小RNA(miRNA)作为转录后调节因子发挥着关键作用,它们是一类短的非编码RNA,来源于被称为前体微小RNA(pre-miRNA)的较长转录本。识别pre-miRNA的实验方法既昂贵又耗时,这就需要计算替代方法。近年来,预测pre-miRNA的计算方法的准确性有了显著提高。然而,仍然存在一些缺点。首先,这些方法通常只考虑碱基频率或序列信息,而忽略了碱基之间的信息。其次,基于二级结构的特征提取方法通常只考虑全局特征,而忽略了局部结构的相互影响。第三,整合高维特征信息的方法计算效率低下。在本研究中,我们提出了一种基于互信息的pre-miRNA序列和二级结构的新型特征表示算法,该算法能够捕捉序列碱基之间的相互作用以及RNA二级结构的局部特征。此外,特征空间比大多数流行方法的特征空间小,这使得我们的方法在计算上比竞争对手更有效。最后,我们应用这些特征训练了一个支持向量机模型来预测pre-miRNA,并将结果与其他流行的预测器进行比较。结果表明,在5折交叉验证和留一法检验中,我们的方法均优于其他方法。