Ali Ameena M, Atmaj Jack, Van Oosterwijk Niels, Groves Matthew R, Dömling Alexander
Department of Drug Design, University of Groningen, Antonius Deusinglaan1, 9700AD Groningen, the Netherlands.
Comput Struct Biotechnol J. 2019 Feb 19;17:263-281. doi: 10.1016/j.csbj.2019.01.012. eCollection 2019.
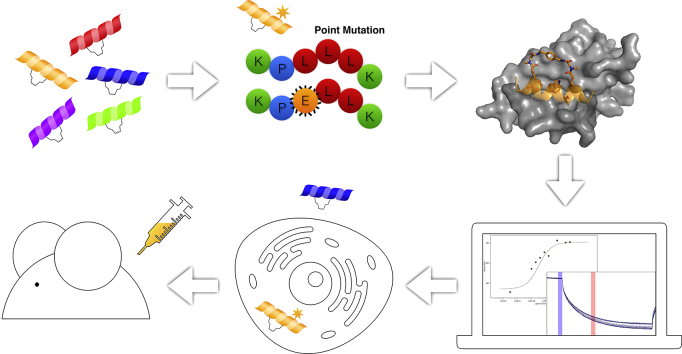
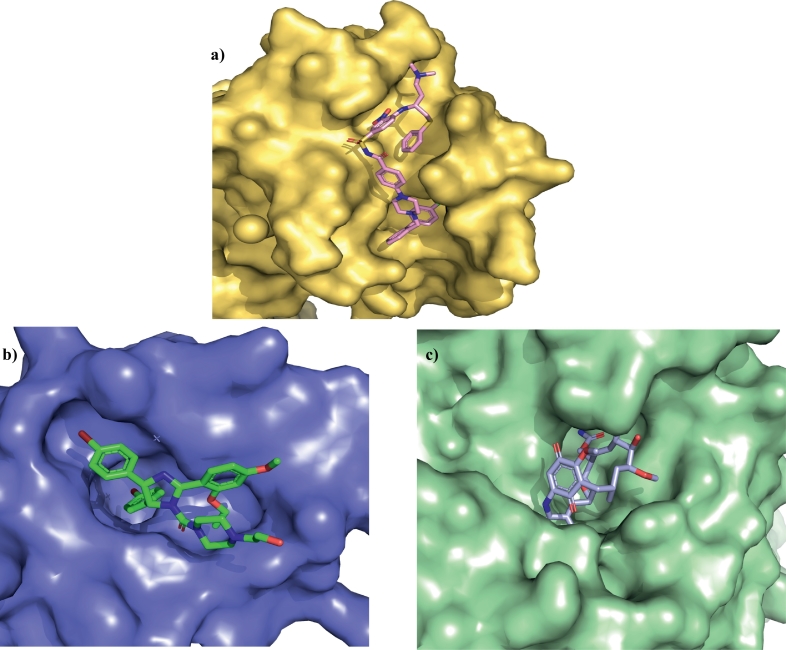
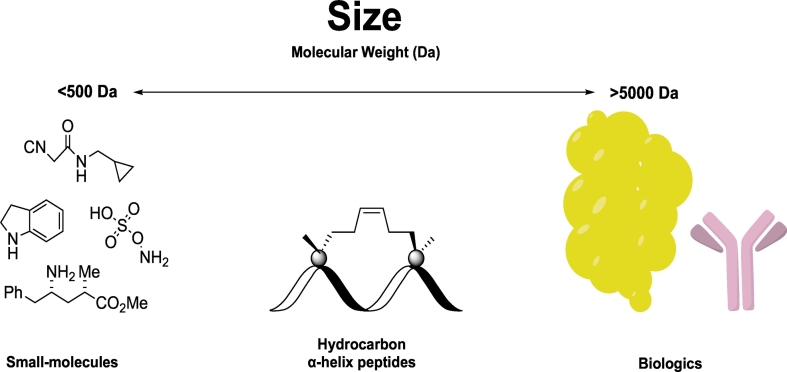
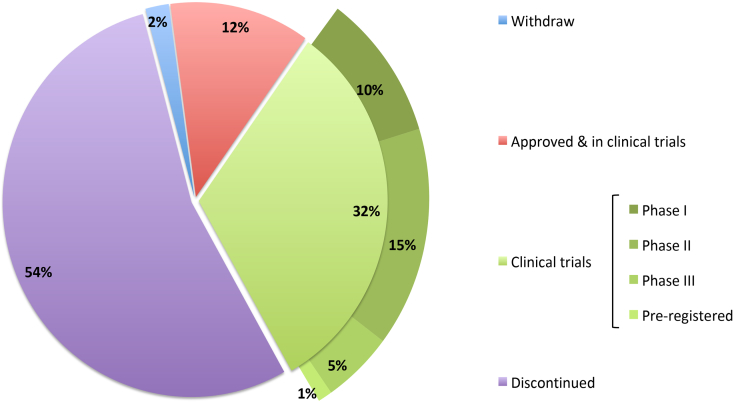
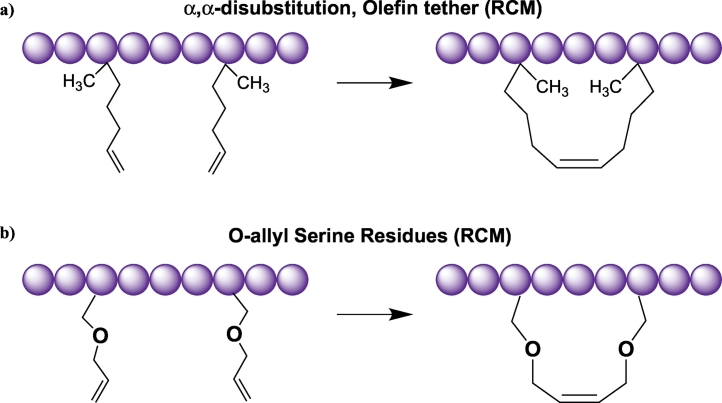
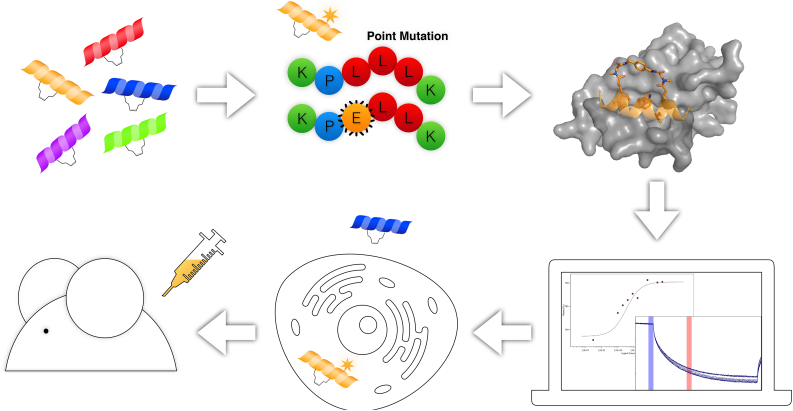
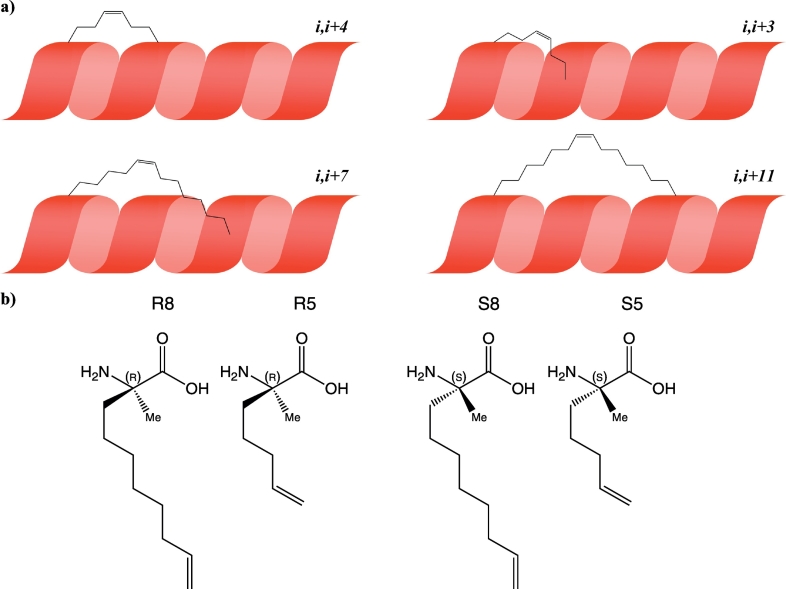
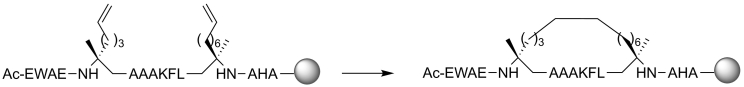
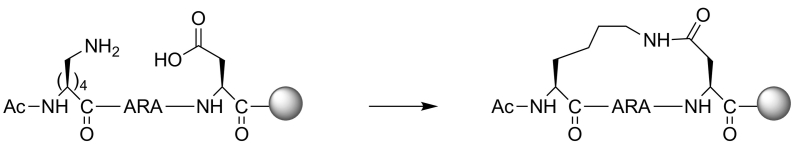
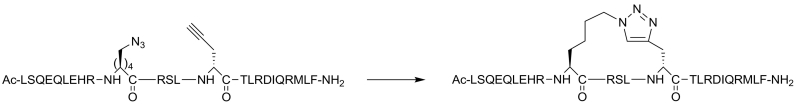
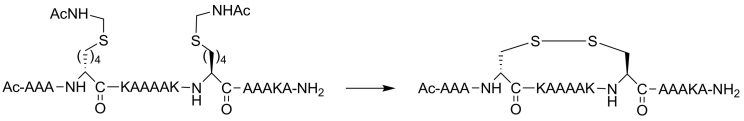
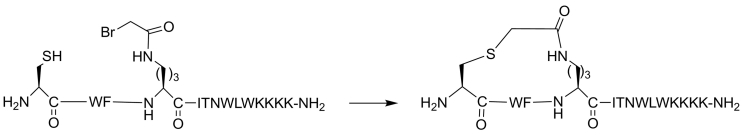
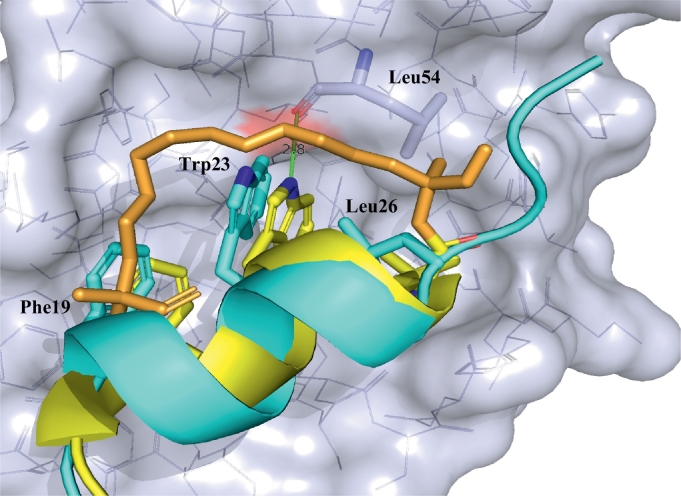
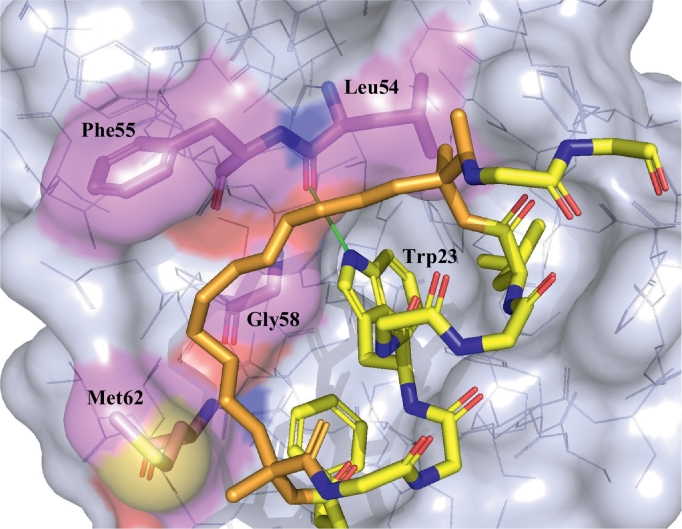
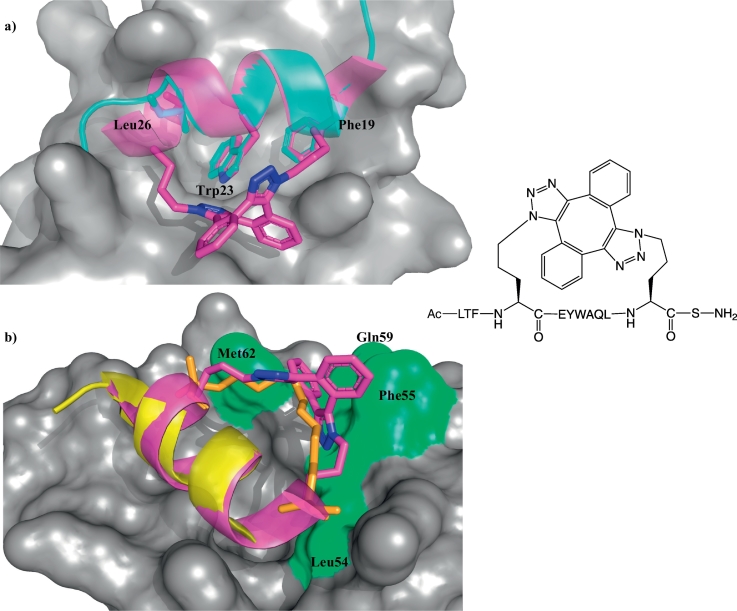
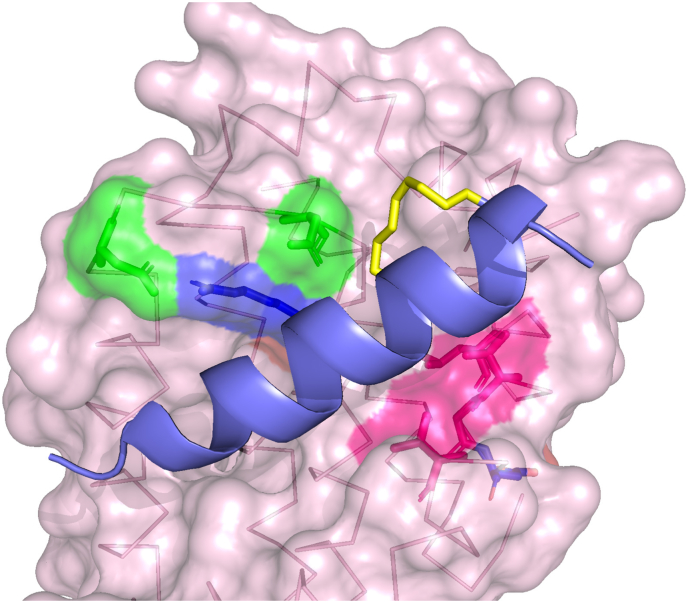
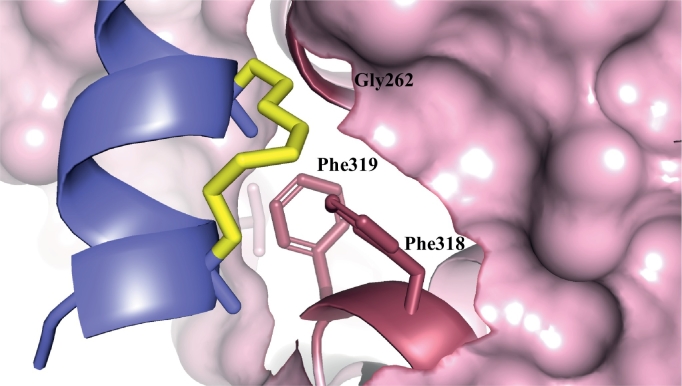
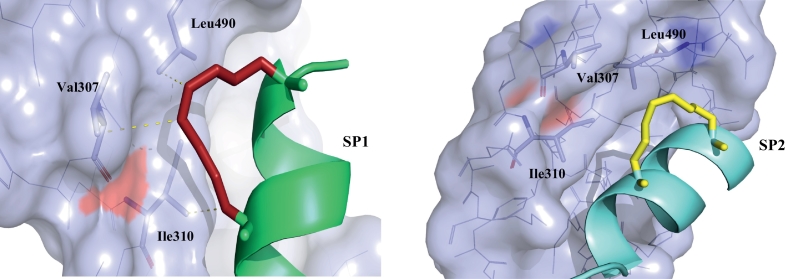
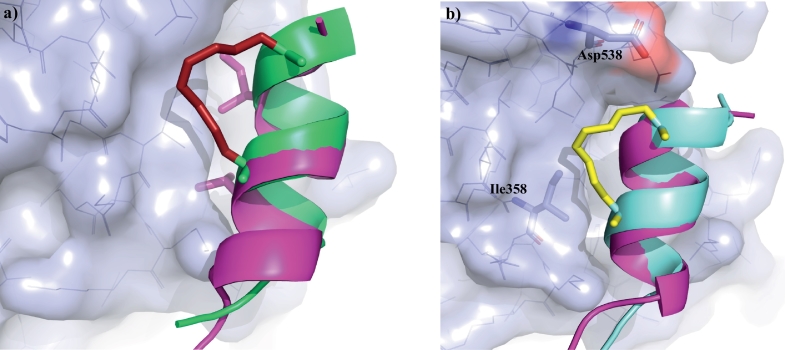
Protein-protein interaction (PPI) is a hot topic in clinical research as protein networking has a major impact in human disease. Such PPIs are potential drugs targets, leading to the need to inhibit/block specific PPIs. While small molecule inhibitors have had some success and reached clinical trials, they have generally failed to address the flat and large nature of PPI surfaces. As a result, larger biologics were developed for PPI surfaces and they have successfully targeted PPIs located outside the cell. However, biologics have low bioavailability and cannot reach intracellular targets. A novel class -hydrocarbon-stapled α-helical peptides that are synthetic mini-proteins locked into their bioactive structure through site-specific introduction of a chemical linker- has shown promise. Stapled peptides show an ability to inhibit intracellular PPIs that previously have been intractable with traditional small molecule or biologics, suggesting that they offer a novel therapeutic modality. In this review, we highlight what stapling adds to natural-mimicking peptides, describe the revolution of synthetic chemistry techniques and how current drug discovery approaches have been adapted to stabilize active peptide conformations, including ring-closing metathesis (RCM), lactamisation, cycloadditions and reversible reactions. We provide an overview on the available stapled peptide high-resolution structures in the protein data bank, with four selected structures discussed in details due to remarkable interactions of their staple with the target surface. We believe that stapled peptides are promising drug candidates and open the doors for peptide therapeutics to reach currently "undruggable" space.
蛋白质-蛋白质相互作用(PPI)是临床研究中的一个热门话题,因为蛋白质网络在人类疾病中具有重大影响。此类PPI是潜在的药物靶点,因此需要抑制/阻断特定的PPI。虽然小分子抑制剂已取得一些成功并进入临床试验,但它们通常无法解决PPI表面平坦且较大的问题。因此,针对PPI表面开发了更大的生物制剂,并且它们已成功靶向位于细胞外的PPI。然而,生物制剂的生物利用度低,无法到达细胞内靶点。一类新型的——烃链订书钉式α-螺旋肽,即通过位点特异性引入化学连接体锁定在其生物活性结构中的合成微型蛋白质——已显示出前景。订书钉肽显示出抑制细胞内PPI的能力,而这些PPI以前用传统小分子或生物制剂难以处理,这表明它们提供了一种新的治疗方式。在这篇综述中,我们强调了订书钉给天然模拟肽带来的作用,描述了合成化学技术的变革以及当前药物发现方法如何被用于稳定活性肽构象,包括关环复分解反应(RCM)、内酰胺化、环加成反应和可逆反应。我们概述了蛋白质数据库中现有的订书钉肽高分辨率结构,并详细讨论了四个选定的结构,因为它们的订书钉与靶表面有显著的相互作用。我们相信订书钉肽是有前景的候选药物,并为肽类疗法进入目前的“不可成药”领域打开了大门。