Division of Genetics and Cell Biology, San Raffaele Scientific Institute, Milan, Italy.
Università Vita-Salute San Raffaele, Milan, Italy.
Elife. 2019 Mar 14;8:e41168. doi: 10.7554/eLife.41168.
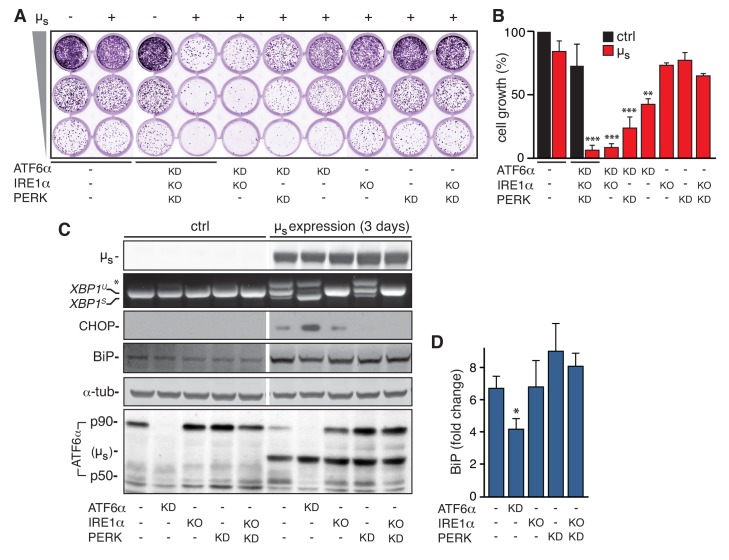
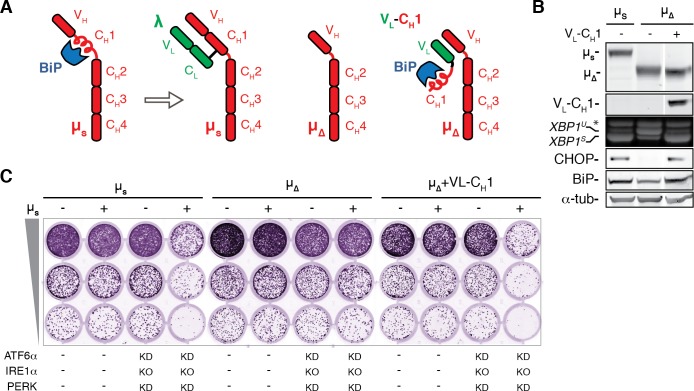
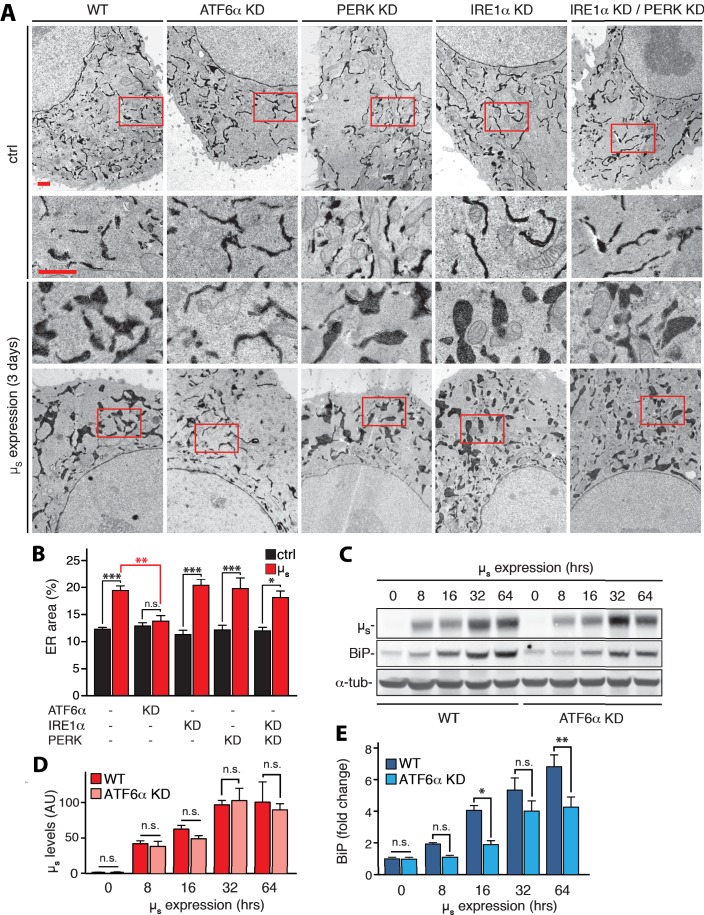
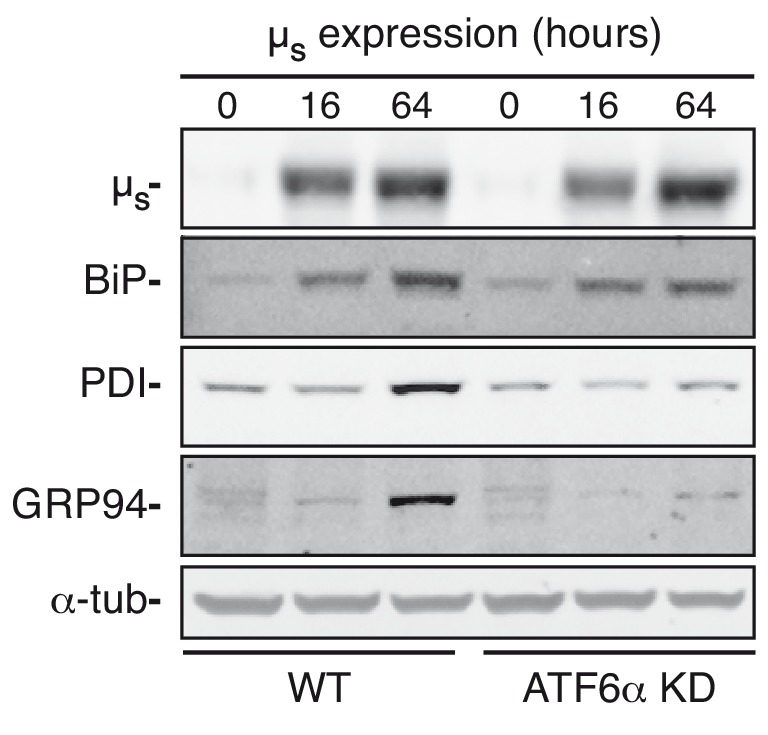
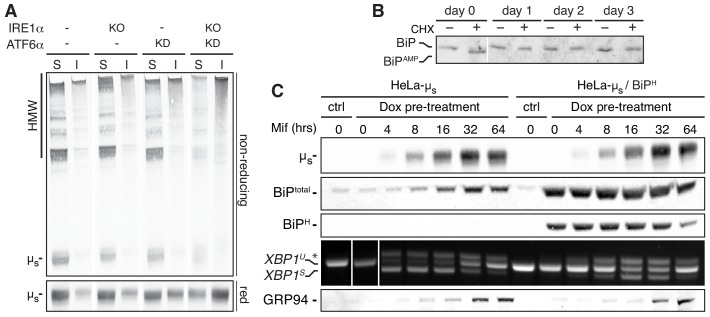
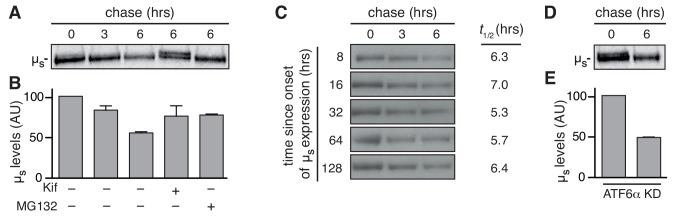
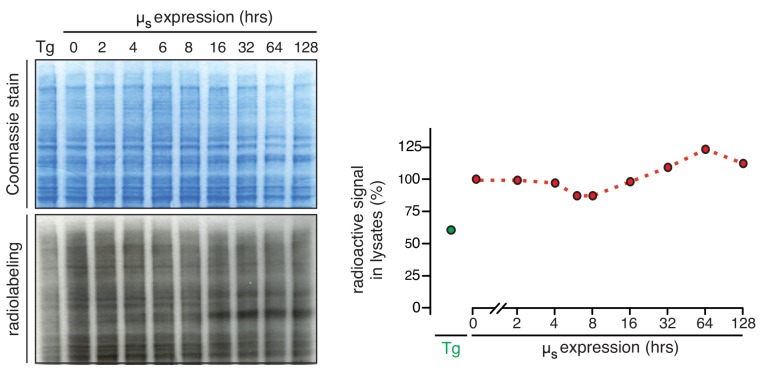
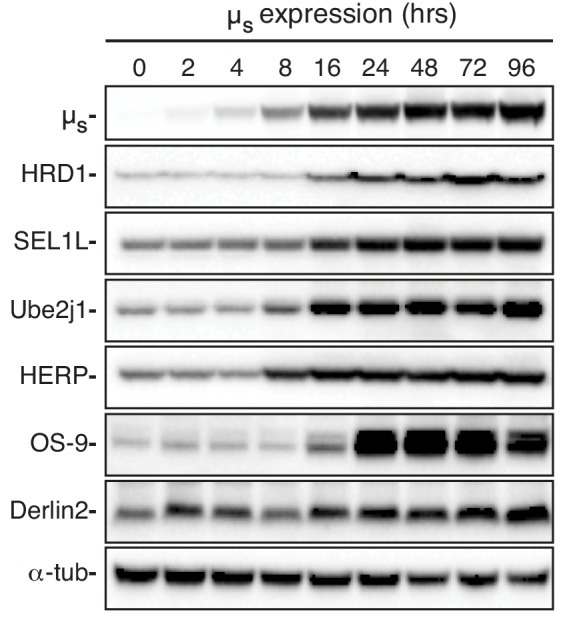
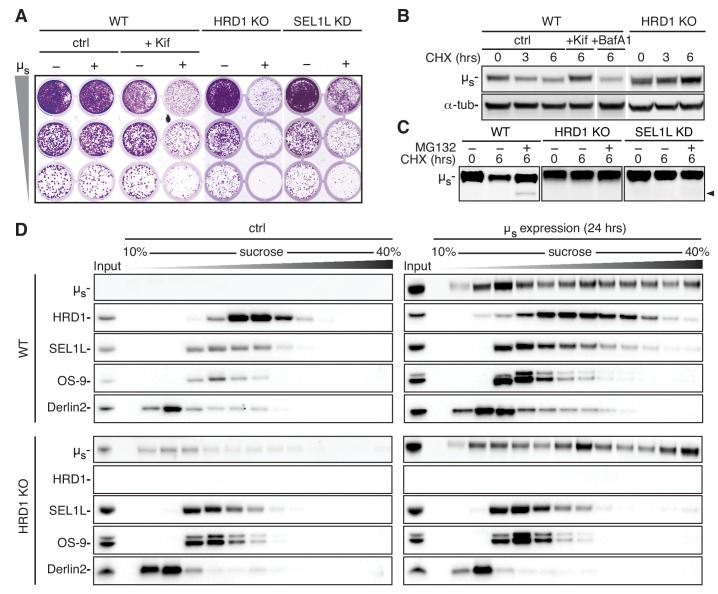
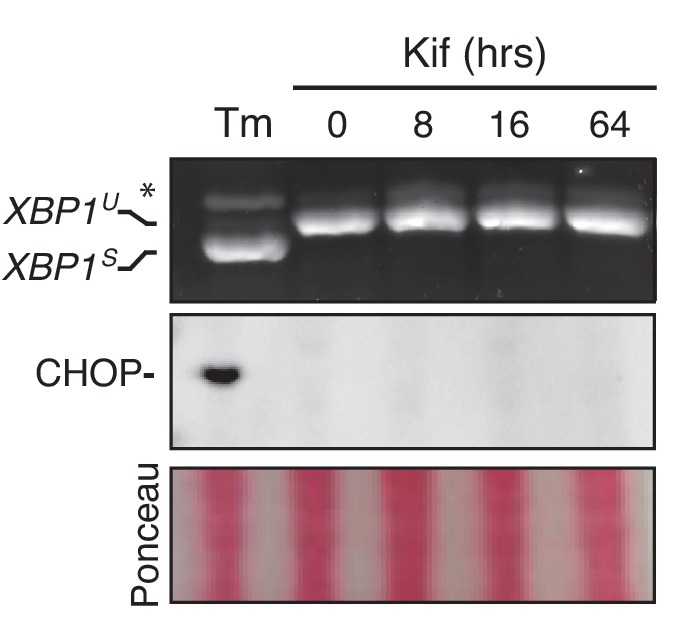
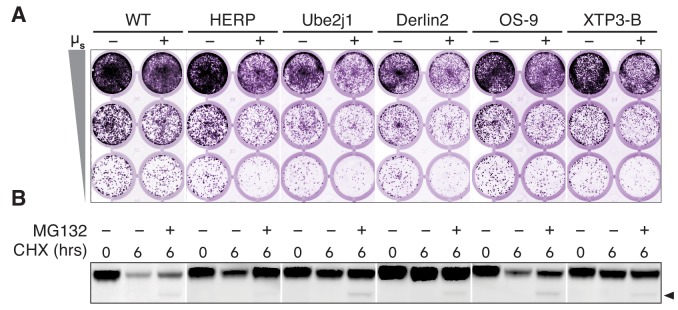
How endoplasmic reticulum (ER) stress leads to cytotoxicity is ill-defined. Previously we showed that HeLa cells readjust homeostasis upon proteostatically driven ER stress, triggered by inducible bulk expression of secretory immunoglobulin M heavy chain (μ) thanks to the unfolded protein response (UPR; Bakunts et al., 2017). Here we show that conditions that prevent that an excess of the ER resident chaperone (and UPR target gene) BiP over µ is restored lead to µ-driven proteotoxicity, i.e. abrogation of HRD1-mediated ER-associated degradation (ERAD), or of the UPR, in particular the ATF6α branch. Such conditions are tolerated instead upon removal of the BiP-sequestering first constant domain (C1) from µ. Thus, our data define proteostatic ER stress to be a specific consequence of inadequate BiP availability, which both the UPR and ERAD redeem.
内质网(ER)应激如何导致细胞毒性尚不清楚。我们之前曾表明,HeLa 细胞在受到未折叠蛋白反应(UPR;Bakunts 等人,2017)触发的由诱导的大量分泌免疫球蛋白 M 重链(μ)表达引起的蛋白稳态驱动的 ER 应激下重新调整了体内平衡。在这里,我们表明,防止 ER 驻留伴侣(和 UPR 靶基因)BiP 超过μ的过量恢复的条件会导致μ驱动的蛋白毒性,即 HRD1 介导的 ER 相关降解(ERAD)的中止,或 UPR 特别是 ATF6α 分支的中止。而当从μ中去除 BiP 结合的第一个恒定结构域(C1)时,则可以耐受这些条件。因此,我们的数据定义了蛋白稳态 ER 应激是 BiP 可用性不足的特定后果,UPR 和 ERAD 都可以缓解该后果。