Jankovska Eliska, Svitek Marek, Holada Karel, Petrak Jiri
1BIOCEV, First Faculty of Medicine, Charles University, Prague, Czech Republic.
2Department of Anesthesiology and Intensive Care, First Faculty of Medicine, Charles University, Prague, Czech Republic.
Clin Proteomics. 2019 Feb 26;16:9. doi: 10.1186/s12014-019-9229-1. eCollection 2019.
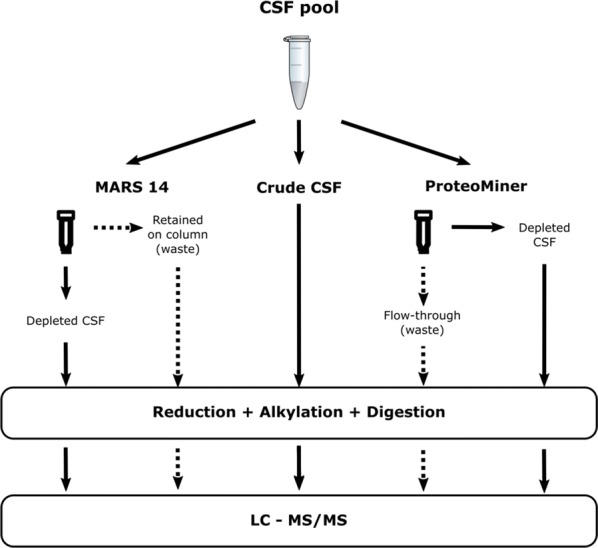
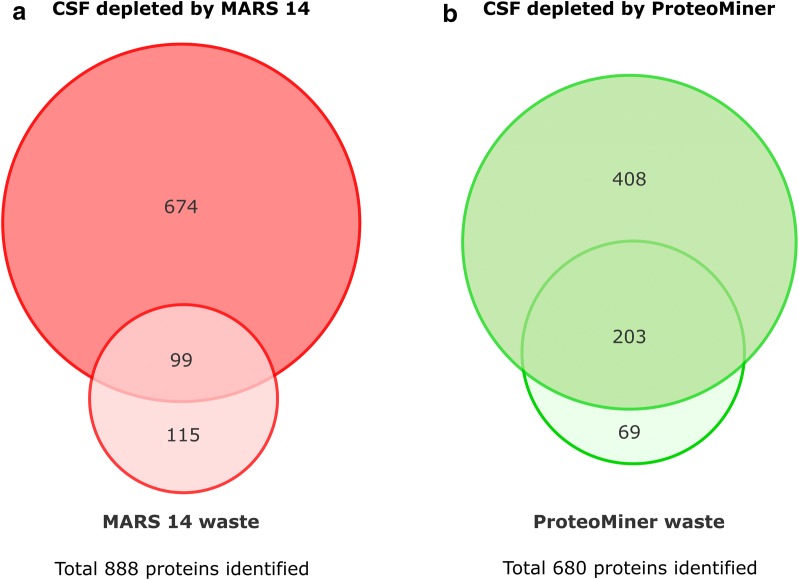
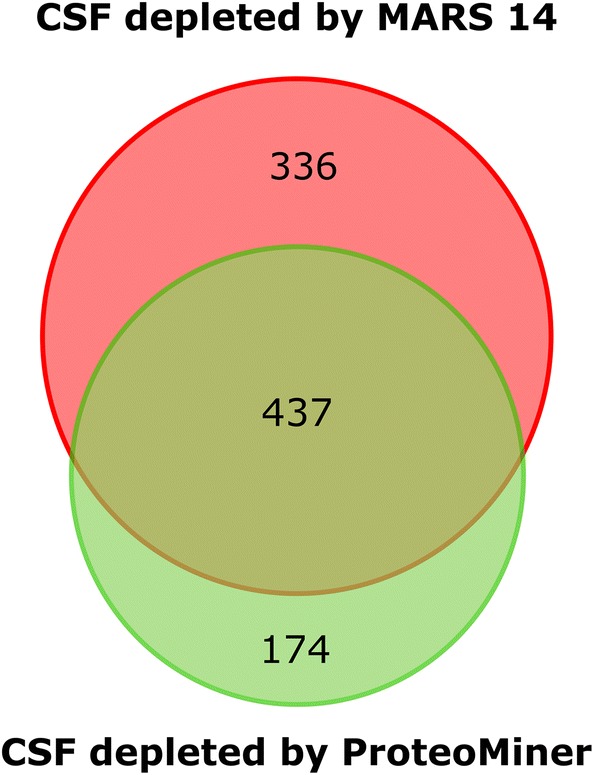
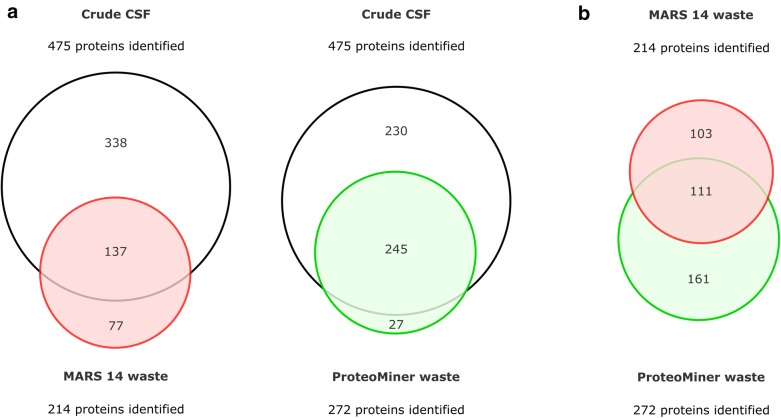
Cerebrospinal fluid (CSF) is in direct contact with the central nervous system. This makes human CSF an attractive source of potential biomarkers for neurologic diseases. Similarly to blood plasma, proteomic analysis of CSF is complicated by a high dynamic range of individual protein concentrations and by the presence of several highly abundant proteins. To deal with the abundant human CSF proteins, methods developed for blood plasma/serum are routinely used. Multiple affinity removal systems and protein enrichment of less abundant proteins using a combinatorial peptide ligand library are among the most frequent approaches. However, their relative impact on CSF proteome coverage has never been evaluated side-by-side in a single study. Therefore, we explored the effect of CSF depletion using MARS 14 cartridge and ProteoMiner ligand library on the number of CSF proteins identified in subsequent LC-MS/MS analysis. LC-MS/MS analysis of crude (non-treated) CSF provided roughly 500 identified proteins. Depletion of CSF by MARS 14 cartridge increased the number of identifications to nearly 800, while treatment of CSF using ProteoMiner enabled identification of 600 proteins. To explore the potential losses of CSF proteins during the depletion process, we also analyzed the "waste" fractions generated by both methods, i.e., proteins retained by the MARS 14 cartridge, and the molecules present in the flow-through fraction from ProteoMiner. More than 250 proteins were bound to MARS 14 cartridge, 100 of those were not identified in the corresponding depleted CSF. Similarly, analysis of the waste fraction in ProteoMiner workflow provided almost 70 unique proteins not found in the CSF depleted by the ligand library. Both depletion strategies significantly increased the number of identified CSF proteins compared to crude CSF. However, MARS 14 depletion provided a markedly higher number of identified proteins (773) compared to ProteoMiner (611). Further, we showed that CSF proteins are lost due to co-depletion (MARS 14) or exclusion (ProteoMiner) during the depletion process. This suggests that the routinely discarded "waste" fractions contain proteins of potential interest and should be included in CSF biomarker studies.
脑脊液(CSF)与中枢神经系统直接接触。这使得人类脑脊液成为神经系统疾病潜在生物标志物的一个有吸引力的来源。与血浆类似,脑脊液的蛋白质组分析因个体蛋白质浓度的高动态范围以及几种高丰度蛋白质的存在而变得复杂。为了处理脑脊液中丰富的蛋白质,通常使用为血浆/血清开发的方法。多种亲和去除系统以及使用组合肽配体库对低丰度蛋白质进行蛋白质富集是最常用的方法。然而,它们对脑脊液蛋白质组覆盖范围的相对影响从未在一项研究中进行过并列评估。因此,我们探讨了使用MARS 14柱和ProteoMiner配体库去除脑脊液对后续液相色谱 - 串联质谱(LC - MS/MS)分析中鉴定出的脑脊液蛋白质数量的影响。对未处理的原始脑脊液进行LC - MS/MS分析大约鉴定出500种蛋白质。使用MARS 14柱去除脑脊液后,鉴定数量增加到近800种,而使用ProteoMiner处理脑脊液能够鉴定出600种蛋白质。为了探究在去除过程中脑脊液蛋白质的潜在损失,我们还分析了两种方法产生的“废液”部分,即MARS 14柱保留的蛋白质以及ProteoMiner流通部分中存在的分子。超过250种蛋白质与MARS 14柱结合,其中100种在相应的去除脑脊液中未被鉴定出来。同样,对ProteoMiner工作流程中的废液部分进行分析,发现了近70种在配体库去除的脑脊液中未发现的独特蛋白质。与原始脑脊液相比,两种去除策略都显著增加了鉴定出的脑脊液蛋白质数量。然而,与ProteoMiner(611种)相比,MARS 14去除提供了明显更多的鉴定蛋白质(773种)。此外,我们表明在去除过程中,脑脊液蛋白质由于共去除(MARS 14)或排除(ProteoMiner)而损失。这表明常规丢弃的“废液”部分包含潜在有意义的蛋白质,应纳入脑脊液生物标志物研究中。