Department of Epidemiology and Biostatistics, Michigan State University, East Lansing, MI 48824
Institute for Quantitative Health Science and Engineering, Michigan State University, East Lansing, MI 48824.
G3 (Bethesda). 2019 May 7;9(5):1377-1383. doi: 10.1534/g3.119.400018.
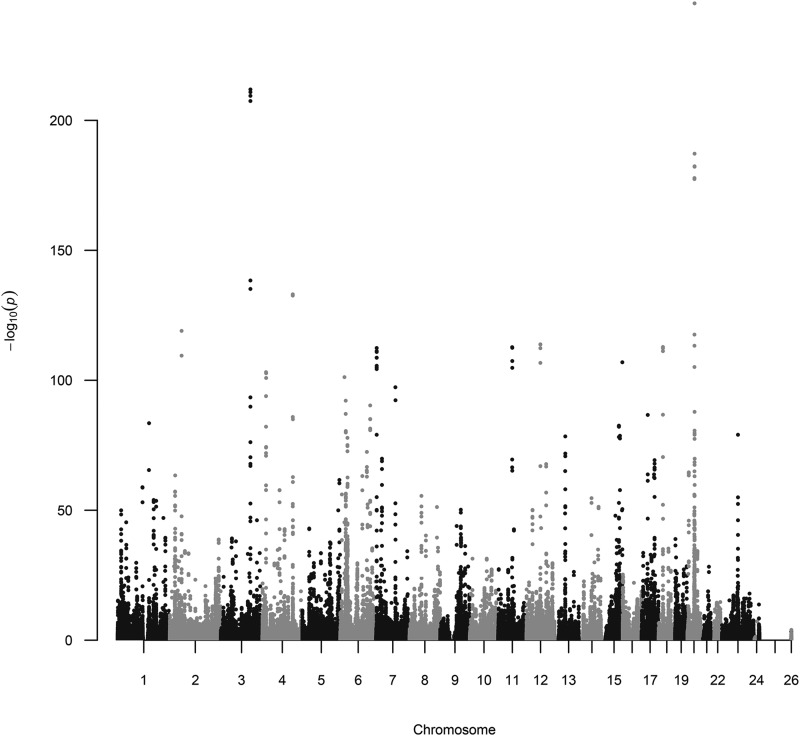
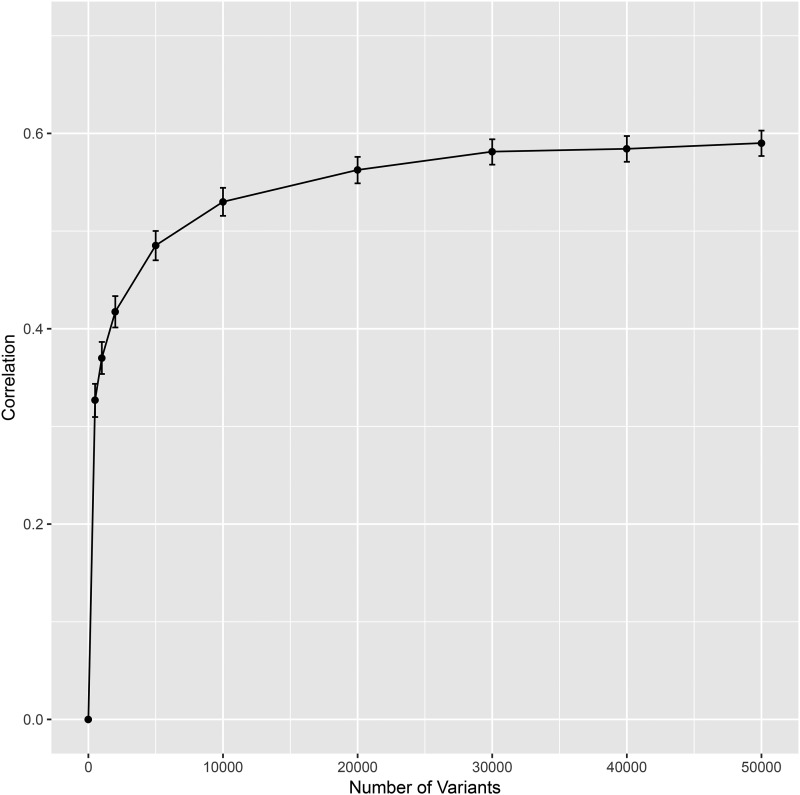
We created a suite of packages to enable analysis of extremely large genomic data sets (potentially millions of individuals and millions of molecular markers) within the R environment. The package offers: a matrix-like interface for .bed files (PLINK's binary format for genotype data), a novel class of linked arrays that allows linking data stored in multiple files to form a single array accessible from the R computing environment, methods for parallel computing capabilities that can carry out computations on very large data sets without loading the entire data into memory and a basic set of methods for statistical genetic analyses. The package is accessible through CRAN and GitHub. In this note, we describe the classes and methods implemented in each of the packages that make the suite and illustrate the use of the packages using data from the UK Biobank.
我们创建了一套软件包,可在 R 环境中对非常大的基因组数据集(可能有数百万个体和数百万分子标记)进行分析。该软件包提供了:用于.bed 文件(PLINK 的基因型数据二进制格式)的矩阵式接口,一种新的链接数组类,允许将存储在多个文件中的数据链接起来,形成一个可从 R 计算环境访问的单个数组,用于并行计算能力的方法,可以在不将整个数据加载到内存中的情况下对非常大数据集进行计算,以及一组基本的统计遗传分析方法。该软件包可通过 CRAN 和 GitHub 使用。在本说明中,我们描述了构成该套件的每个软件包中实现的类和方法,并使用来自 UK Biobank 的数据说明了软件包的使用。