Mater Research Institute - The University of Queensland, Translational Research Institute, Woolloongabba, QLD, Australia.
CIC-IT 1429, Service de Médecine Physique et de Réadaptation, Raymond Poincaré University Hospital, AP-HP, Garches, France.
Front Immunol. 2019 Mar 7;10:377. doi: 10.3389/fimmu.2019.00377. eCollection 2019.
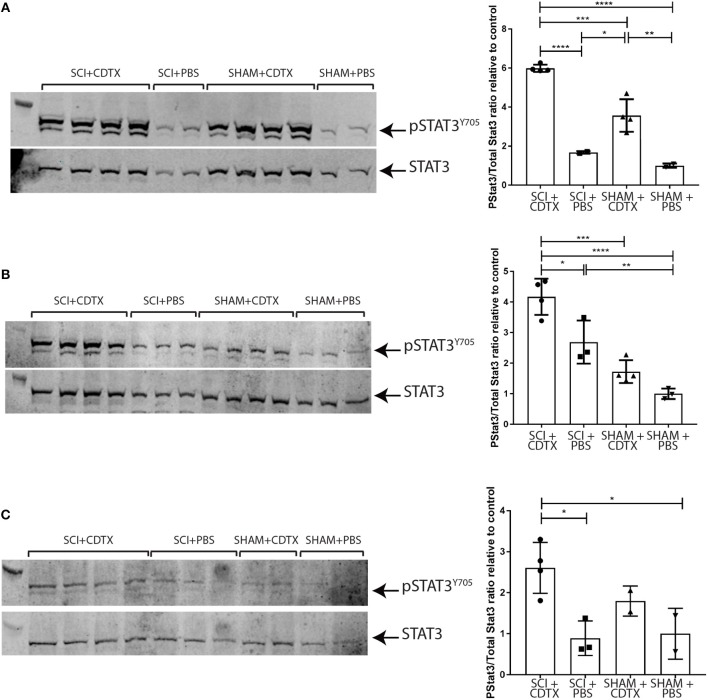
Neurogenic heterotopic ossifications (NHO) are very incapacitating complications of traumatic brain and spinal cord injuries (SCI) which manifest as abnormal formation of bone tissue in periarticular muscles. NHO are debilitating as they cause pain, partial or total joint ankylosis and vascular and nerve compression. NHO pathogenesis is unknown and the only effective treatment remains surgical resection, however once resected, NHO can re-occur. To further understand NHO pathogenesis, we developed the first animal model of NHO following SCI in genetically unmodified mice, which mimics most clinical features of NHO in patients. We have previously shown that the combination of (1) a central nervous system lesion (SCI) and (2) muscular damage (via an intramuscular injection of cardiotoxin) is required for NHO development. Furthermore, macrophages within the injured muscle play a critical role in driving NHO pathogenesis. More recently we demonstrated that macrophage-derived oncostatin M (OSM) is a key mediator of both human and mouse NHO. We now report that inflammatory monocytes infiltrate the injured muscles of SCI mice developing NHO at significantly higher levels compared to mice without SCI. Muscle infiltrating monocytes and neutrophils expressed OSM whereas mouse muscle satellite and interstitial cell expressed the OSM receptor (OSMR). recombinant mouse OSM induced tyrosine phosphorylation of the transcription factor STAT3, a downstream target of OSMR:gp130 signaling in muscle progenitor cells. As STAT3 is tyrosine phosphorylated by JAK1/2 tyrosine kinases downstream of OSMR:gp130, we demonstrated that the JAK1/2 tyrosine kinase inhibitor ruxolitinib blocked OSM driven STAT3 tyrosine phosphorylation in mouse muscle progenitor cells. We further demonstrated that STAT3 tyrosine phosphorylation was not only significantly higher but persisted for a longer duration in injured muscles of SCI mice developing NHO compared to mice with muscle injury without SCI. Finally, administration of ruxolitinib for 7 days post-surgery significantly reduced STAT3 phosphorylation in injured muscles as well as NHO volume at all analyzed time-points up to 3 weeks post-surgery. Our results identify the JAK/STAT3 signaling pathway as a potential therapeutic target to reduce NHO development following SCI.
神经源性异位骨化(NHO)是颅脑和脊髓损伤(SCI)的一种非常致残的并发症,表现为关节周围肌肉中异常骨组织的形成。NHO 会导致疼痛、部分或完全关节强直以及血管和神经受压,因此具有致残性。NHO 的发病机制尚不清楚,唯一有效的治疗方法仍然是手术切除,但一旦切除,NHO 可能会再次发生。为了进一步了解 NHO 的发病机制,我们在未经基因修饰的小鼠中建立了首个 SCI 后 NHO 的动物模型,该模型模拟了患者 NHO 的大多数临床特征。我们之前已经表明,(1)中枢神经系统损伤(SCI)和(2)肌肉损伤(通过肌肉内注射心脏毒素)的结合是 NHO 发展所必需的。此外,受伤肌肉内的巨噬细胞在驱动 NHO 发病机制中起着关键作用。最近,我们证明巨噬细胞衍生的肿瘤坏死因子样细胞因子超家族成员 11(OSM)是人类和小鼠 NHO 的关键介质。我们现在报告称,炎性单核细胞浸润到发生 NHO 的 SCI 小鼠的受伤肌肉中,浸润水平明显高于没有 SCI 的小鼠。肌肉浸润的单核细胞和中性粒细胞表达 OSM,而小鼠肌肉卫星细胞和间质细胞表达 OSM 受体(OSMR)。重组小鼠 OSM 诱导肌肉祖细胞中转录因子 STAT3 的酪氨酸磷酸化,OSMR:gp130 信号的下游靶标。由于 OSMR:gp130 下游的 JAK1/2 酪氨酸激酶使 STAT3 酪氨酸磷酸化,我们证明 JAK1/2 酪氨酸激酶抑制剂鲁索利替尼阻断了小鼠肌肉祖细胞中 OSM 驱动的 STAT3 酪氨酸磷酸化。我们进一步证明,与没有 SCI 的肌肉损伤小鼠相比,发生 NHO 的 SCI 小鼠的受伤肌肉中 STAT3 酪氨酸磷酸化不仅水平显著更高,而且持续时间更长。最后,手术后 7 天给予鲁索利替尼治疗可显著降低手术后所有分析时间点的受伤肌肉中的 STAT3 磷酸化以及 3 周内的 NHO 体积。我们的研究结果确定 JAK/STAT3 信号通路是减少 SCI 后 NHO 发展的潜在治疗靶点。