Department of Physics, Karlsruhe Institute of Technology, Karlsruhe, Germany.
Steinbuch Centre for Computing, Karlsruhe Institute of Technology, Eggenstein-Leopoldshafen, Germany.
PLoS Comput Biol. 2019 Mar 22;15(3):e1006900. doi: 10.1371/journal.pcbi.1006900. eCollection 2019 Mar.
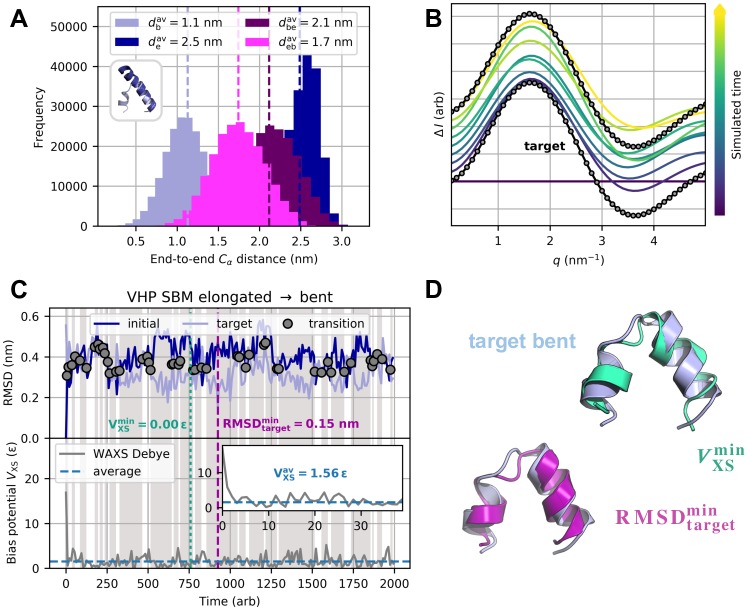
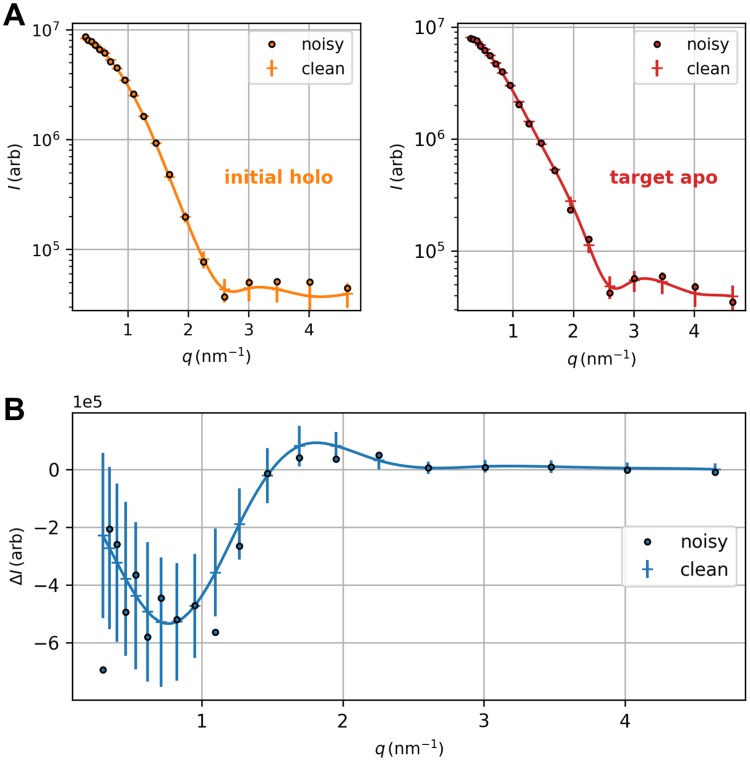
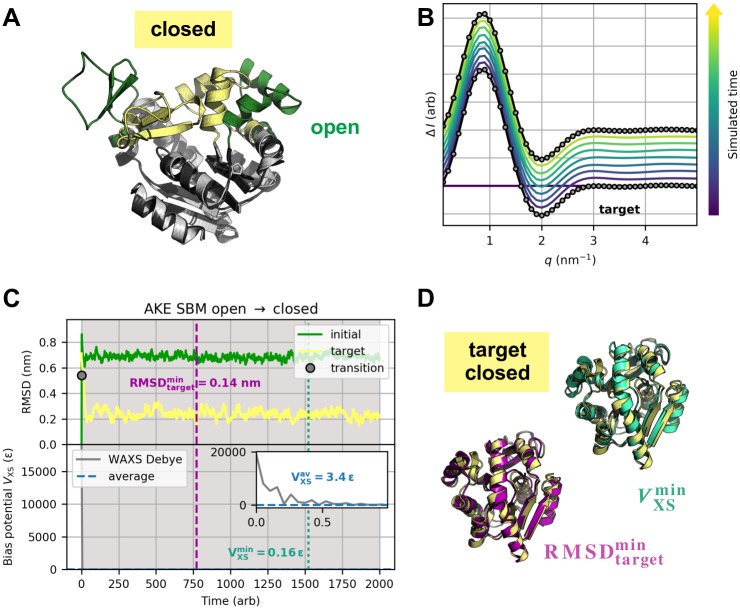
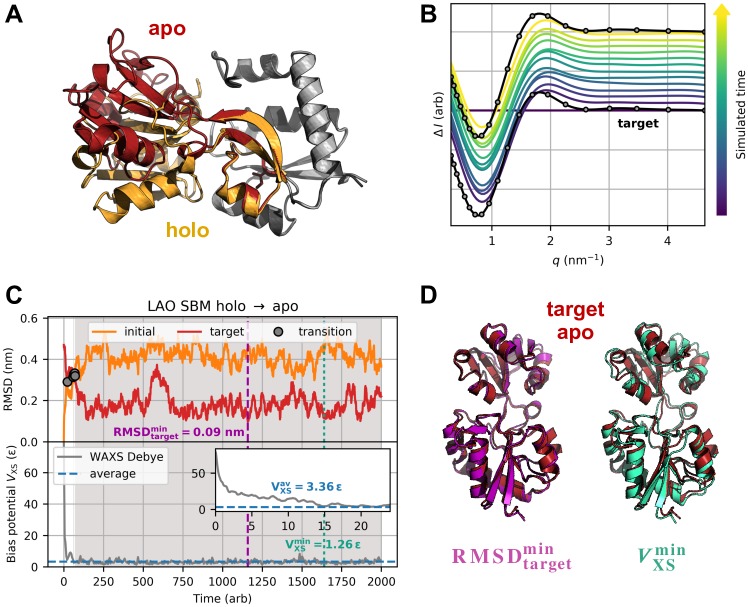
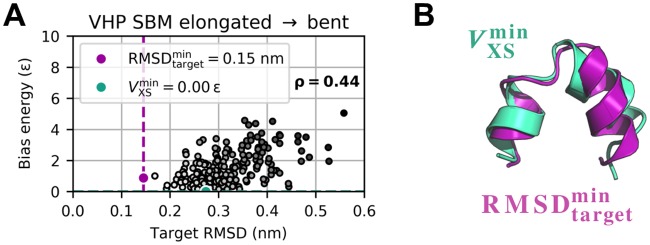
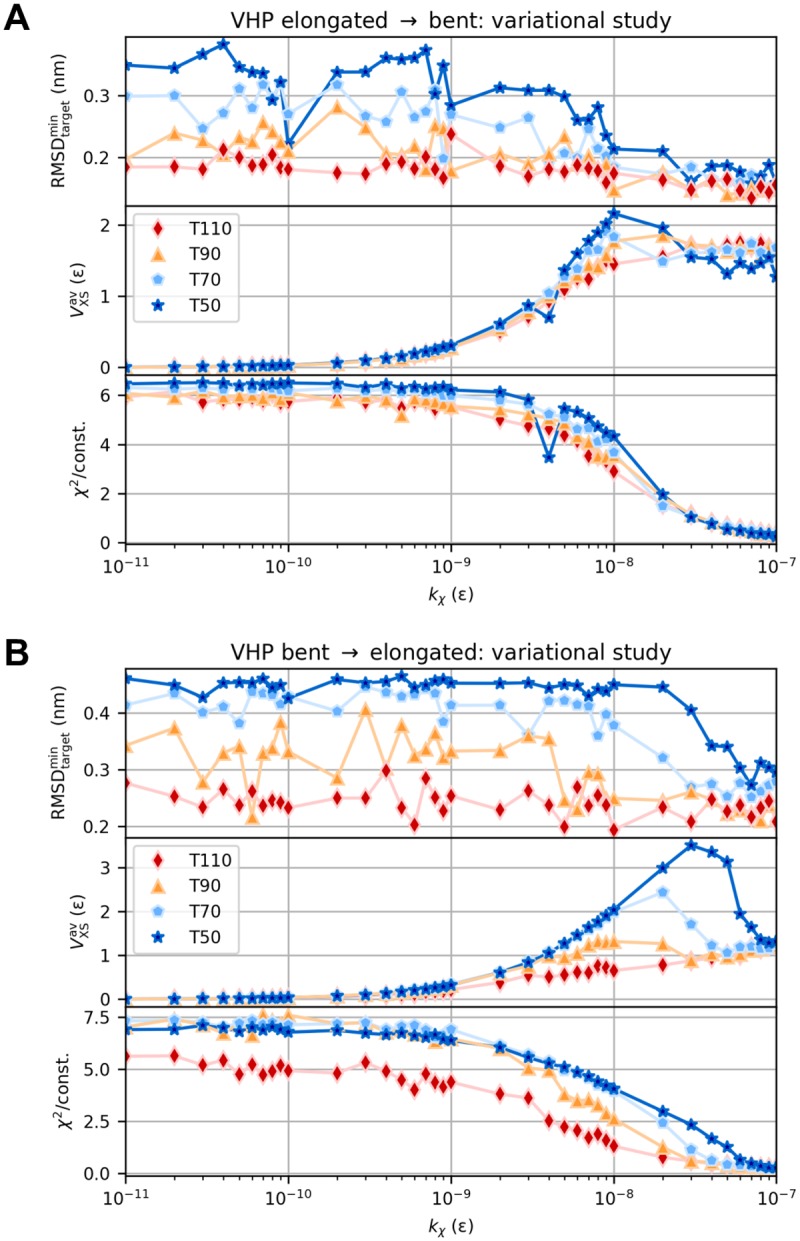
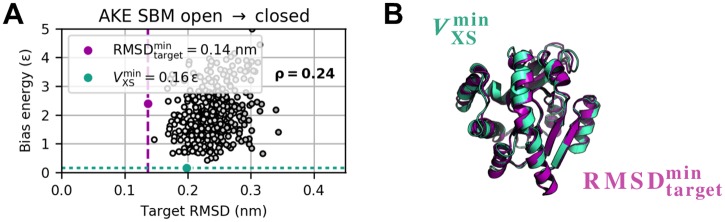
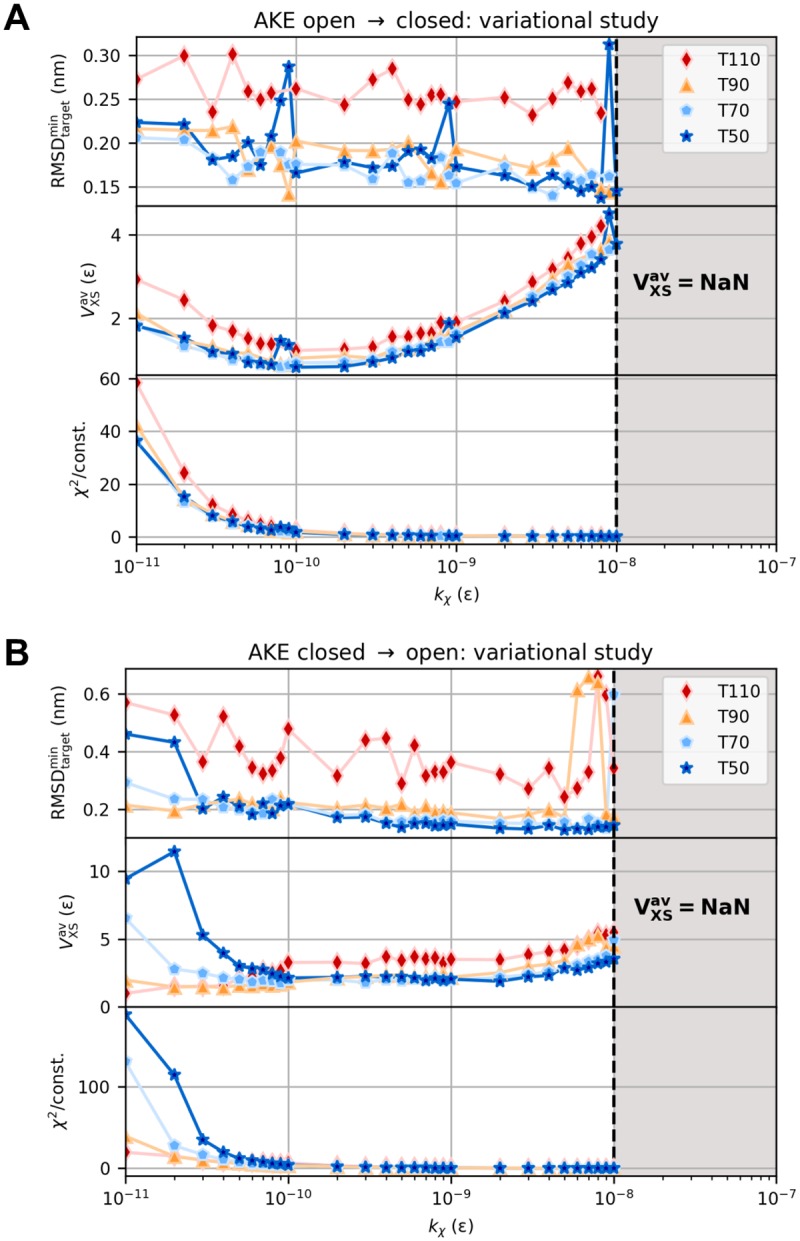
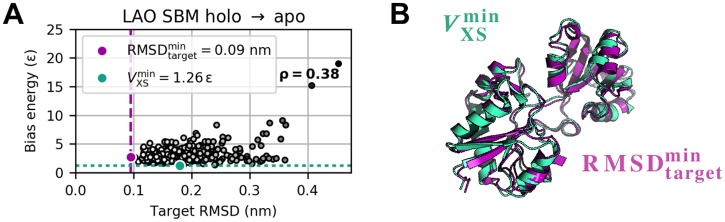
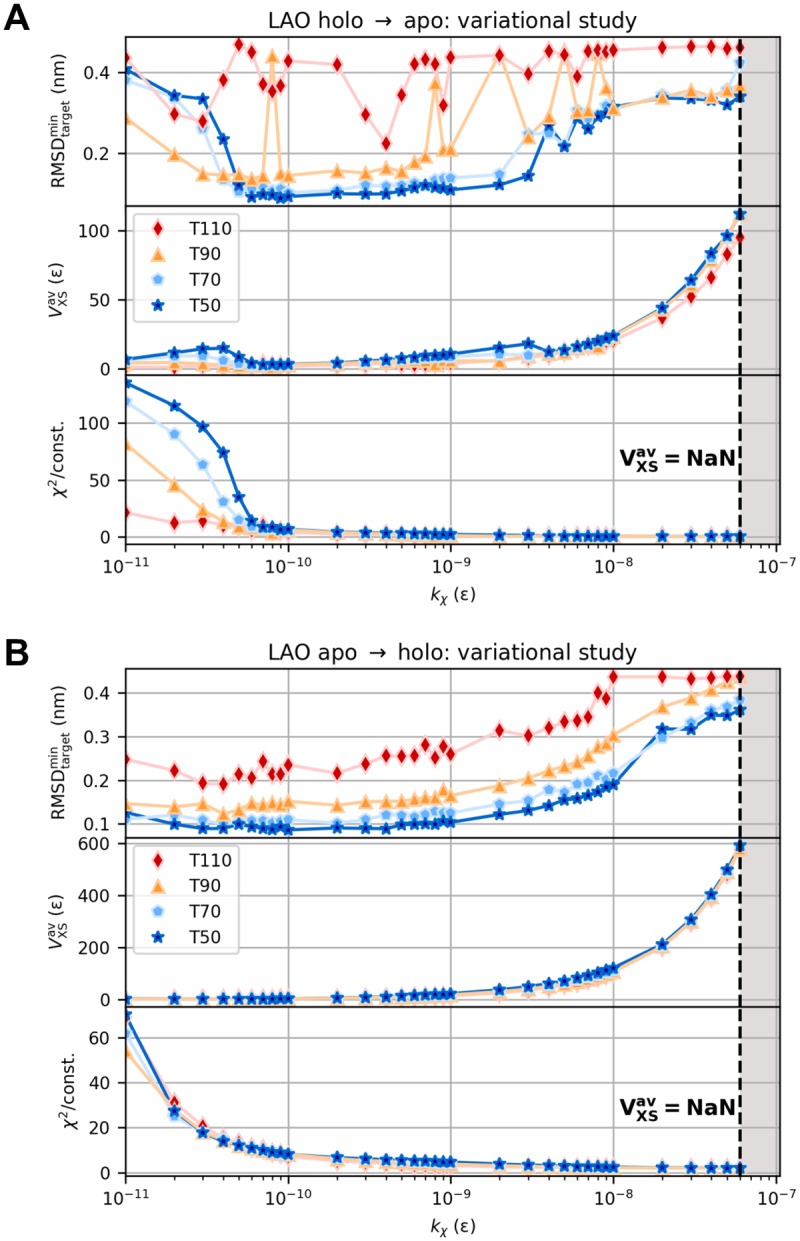
The fundamental aim of structural analyses in biophysics is to reveal a mutual relation between a molecule's dynamic structure and its physiological function. Small-angle X-ray scattering (SAXS) is an experimental technique for structural characterization of macromolecules in solution and enables time-resolved analysis of conformational changes under physiological conditions. As such experiments measure spatially averaged low-resolution scattering intensities only, the sparse information obtained is not sufficient to uniquely reconstruct a three-dimensional atomistic model. Here, we integrate the information from SAXS into molecular dynamics simulations using computationally efficient native structure-based models. Dynamically fitting an initial structure towards a scattering intensity, such simulations produce atomistic models in agreement with the target data. In this way, SAXS data can be rapidly interpreted while retaining physico-chemical knowledge and sampling power of the underlying force field. We demonstrate our method's performance using the example of three protein systems. Simulations are faster than full molecular dynamics approaches by more than two orders of magnitude and consistently achieve comparable accuracy. Computational demands are reduced sufficiently to run the simulations on commodity desktop computers instead of high-performance computing systems. These results underline that scattering-guided structure-based simulations provide a suitable framework for rapid early-stage refinement of structures towards SAXS data with particular focus on minimal computational resources and time.
生物物理学中结构分析的基本目的是揭示分子动态结构与其生理功能之间的相互关系。小角 X 射线散射(SAXS)是一种用于溶液中大分子结构特征的实验技术,能够在生理条件下实时分析构象变化。由于这些实验仅测量空间平均的低分辨率散射强度,因此获得的稀疏信息不足以唯一重建三维原子模型。在这里,我们使用计算效率高的基于天然结构的模型,将 SAXS 的信息整合到分子动力学模拟中。通过动态拟合初始结构与散射强度,这种模拟可以生成与目标数据一致的原子模型。通过这种方式,可以快速解释 SAXS 数据,同时保留物理化学知识和基础力场的采样能力。我们使用三个蛋白质系统的例子来展示我们方法的性能。模拟的速度比全分子动力学方法快两个数量级以上,并且始终达到可比的准确性。计算需求降低到足以在商业台式计算机上而不是高性能计算系统上运行模拟。这些结果表明,散射引导的基于结构的模拟为快速早期阶段的结构精修提供了一个合适的框架,特别关注最小的计算资源和时间。