Legendre Matthieu, Alempic Jean-Marie, Philippe Nadège, Lartigue Audrey, Jeudy Sandra, Poirot Olivier, Ta Ngan Thi, Nin Sébastien, Couté Yohann, Abergel Chantal, Claverie Jean-Michel
Aix Marseille Univ, CNRS, IGS, Structural and Genomic Information Laboratory (UMR7256), Mediterranean Institute of Microbiology (FR3479), Marseille, France.
Inserm, BIG-BGE, CEA, Université Grenoble Alpes, Grenoble, France.
Front Microbiol. 2019 Mar 8;10:430. doi: 10.3389/fmicb.2019.00430. eCollection 2019.
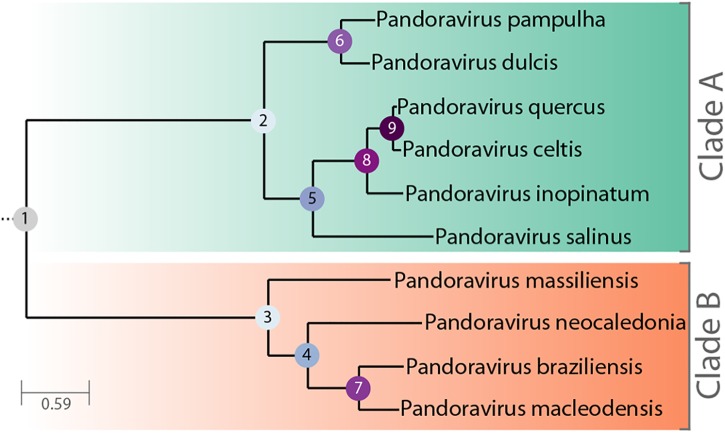
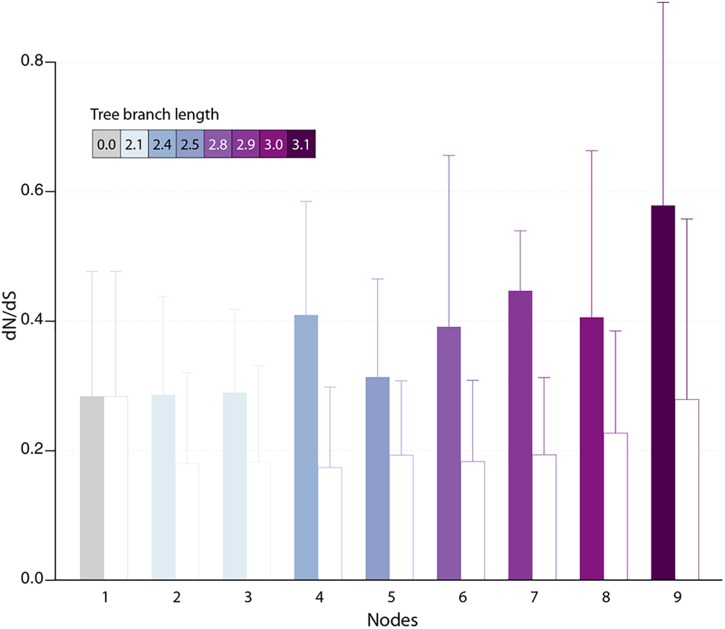
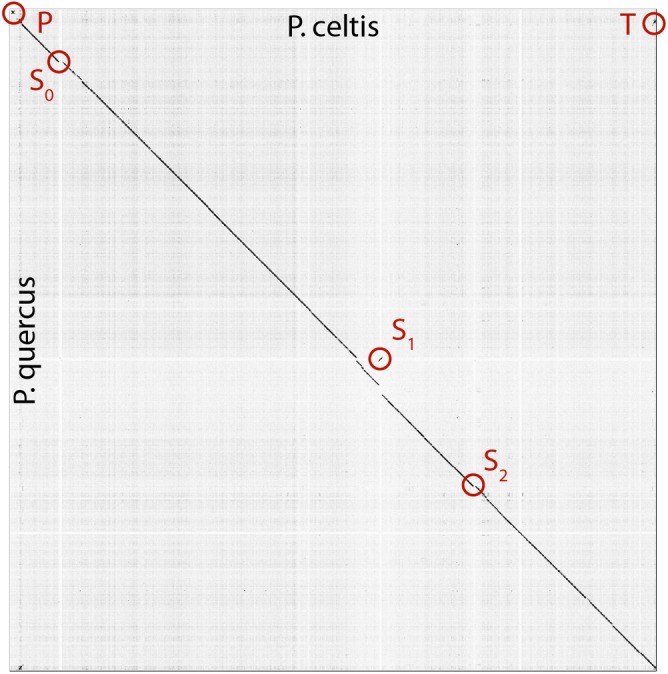
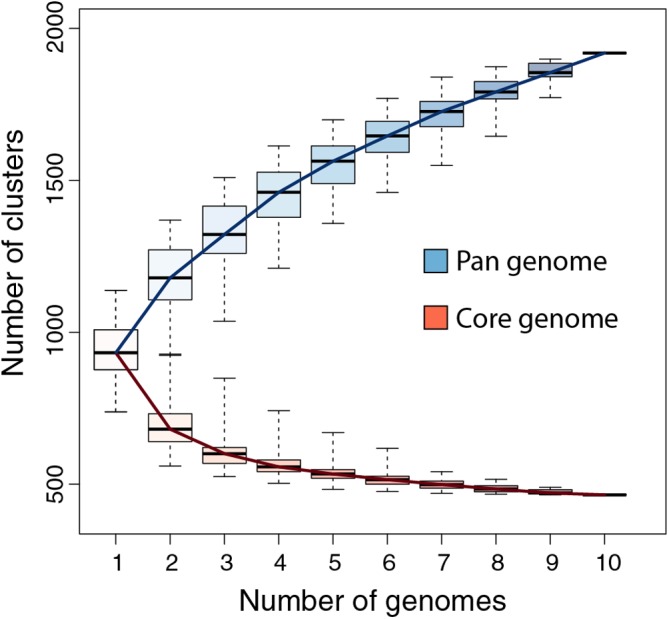
With genomes of up to 2.7 Mb propagated in μm-long oblong particles and initially predicted to encode more than 2000 proteins, members of the family display the most extreme features of the known viral world. The mere existence of such giant viruses raises fundamental questions about their origin and the processes governing their evolution. A previous analysis of six newly available isolates, independently confirmed by a study including three others, established that the pan-genome is open, meaning that each new strain exhibits protein-coding genes not previously identified in other family members. With an average increment of about 60 proteins, the gene repertoire shows no sign of reaching a limit and remains largely coding for proteins without recognizable homologs in other viruses or cells (ORFans). To explain these results, we proposed that most new protein-coding genes were created , from pre-existing non-coding regions of the G+C rich pandoravirus genomes. The comparison of the gene content of a new isolate, pandoravirus celtis, closely related (96% identical genome) to the previously described p. quercus is now used to test this hypothesis by studying genomic changes in a microevolution range. Our results confirm that the differences between these two similar gene contents mostly consist of protein-coding genes without known homologs, with statistical signatures close to that of intergenic regions. These newborn proteins are under slight negative selection, perhaps to maintain stable folds and prevent protein aggregation pending the eventual emergence of fitness-increasing functions. Our study also unraveled several insertion events mediated by a transposase of the hAT family, 3 copies of which are found in p. celtis and are presumably active. Members of the are presently the first viruses known to encode this type of transposase.
该病毒家族的成员在微米长的椭圆形颗粒中传播,基因组可达2.7兆碱基对,最初预计编码超过2000种蛋白质,展现出已知病毒世界中最为极端的特征。如此巨大的病毒的存在本身就引发了关于其起源以及控制其进化过程的基本问题。先前对六个新获得的分离株的分析,经另一项包含其他三个分离株的研究独立证实,确定该病毒家族的泛基因组是开放的,这意味着每个新菌株都具有先前在其他家族成员中未发现的蛋白质编码基因。基因库平均增加约60种蛋白质,没有迹象表明会达到极限,并且很大程度上仍然编码在其他病毒或细胞中没有可识别同源物的蛋白质(孤儿基因)。为了解释这些结果,我们提出大多数新的蛋白质编码基因是由富含G+C的潘多拉病毒基因组中预先存在的非编码区域产生的。现在,通过研究微观进化范围内的基因组变化,比较新分离株潘多拉病毒celtis(与先前描述的潘多拉病毒quercus密切相关,基因组96%相同)的基因内容,来检验这一假设。我们的结果证实,这两个相似基因库之间的差异主要由没有已知同源物的蛋白质编码基因组成,其统计特征与基因间区域相近。这些新生蛋白质受到轻微的负选择,可能是为了维持稳定的折叠结构,并在最终出现增加适应性的功能之前防止蛋白质聚集。我们的研究还揭示了由hAT家族转座酶介导的几个插入事件,在潘多拉病毒celtis中发现了3个该转座酶的拷贝,推测它们是活跃的。潘多拉病毒家族的成员目前是已知的首个编码这种转座酶的病毒。