Aix Marseille Univ, CNRS, Structural and Genomic Information Laboratory, UMR 7256 (IMM FR 3479), 163 Avenue de Luminy, Case 934, 13288, Marseille cedex 9, France.
Univ. Grenoble Alpes, CEA, Inserm, BIG-BGE, 38000, Grenoble, France.
Nat Commun. 2018 Jun 11;9(1):2285. doi: 10.1038/s41467-018-04698-4.
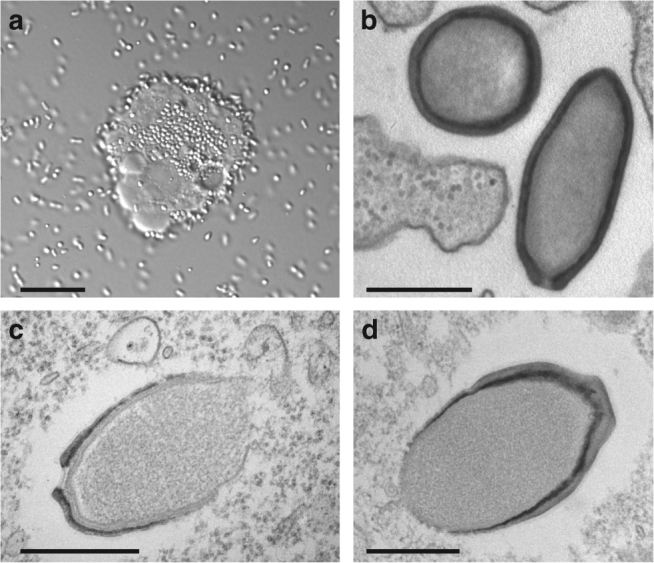
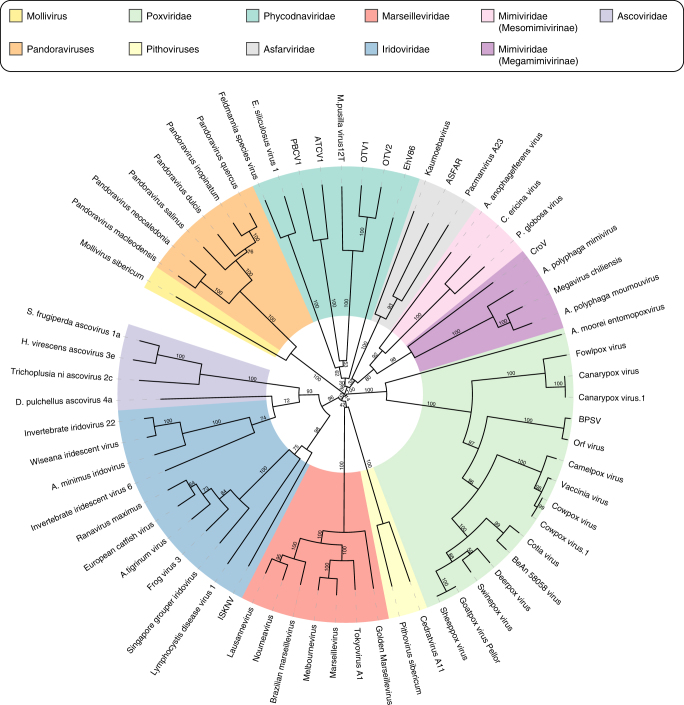
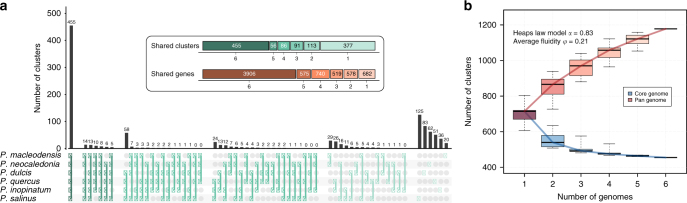
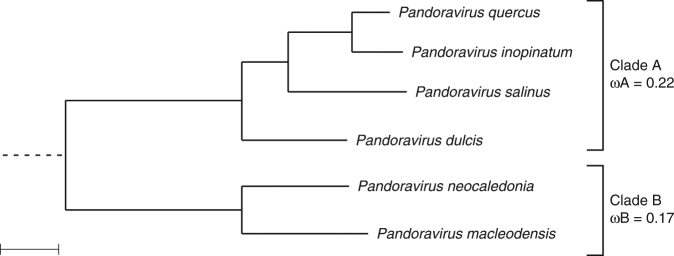
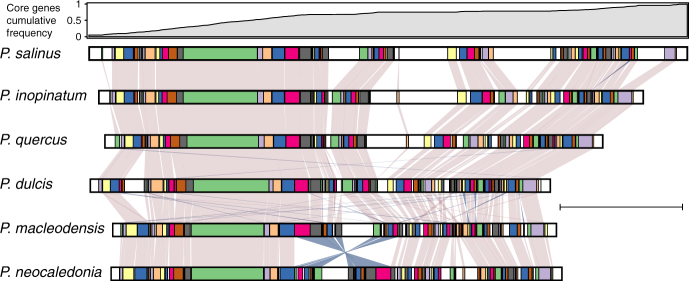
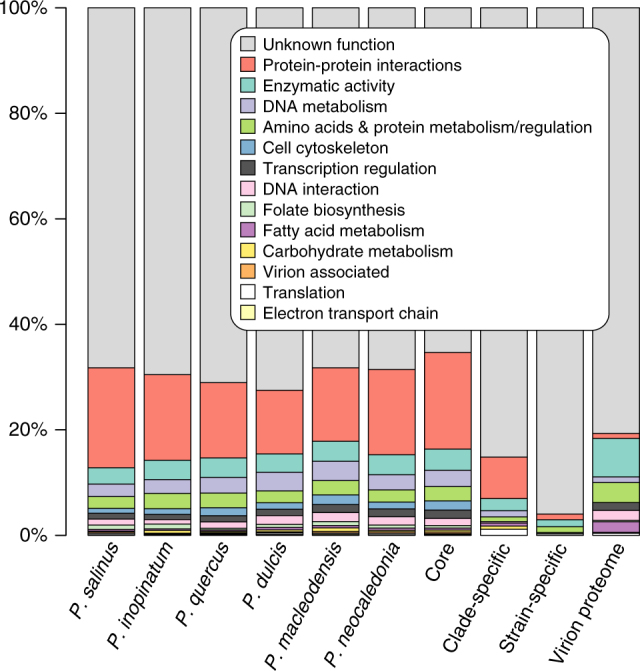
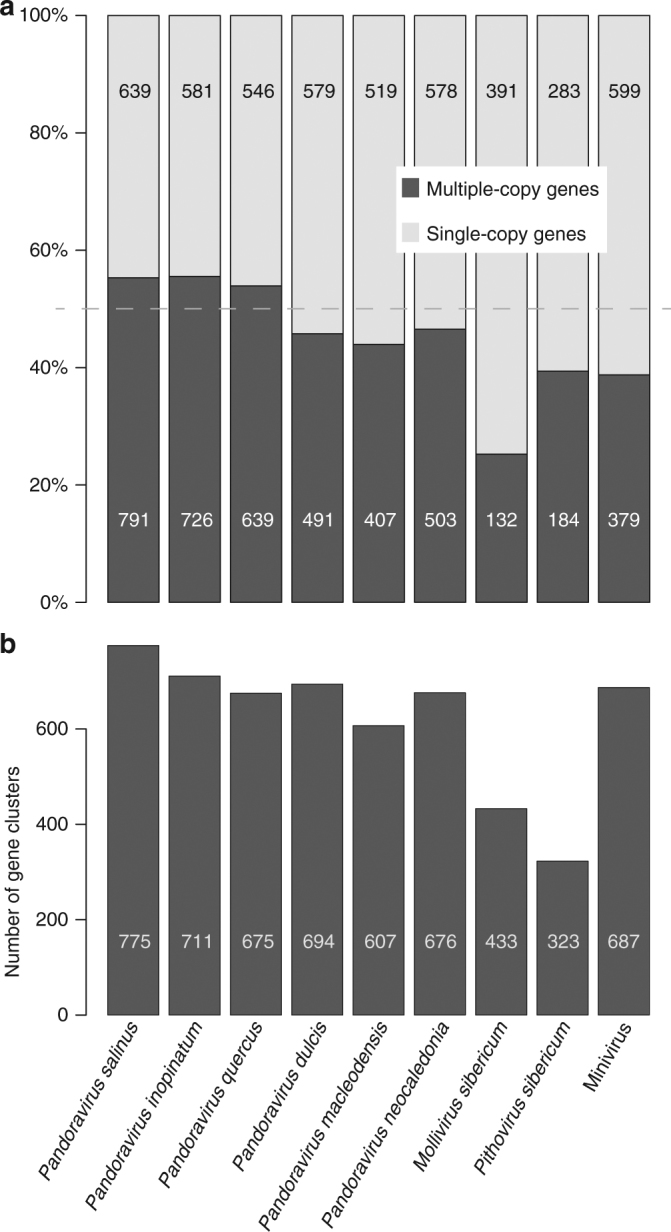
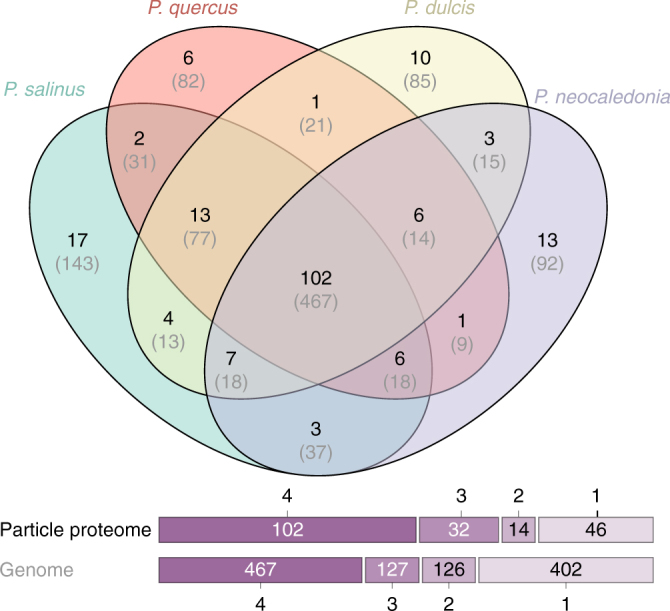
With DNA genomes reaching 2.5 Mb packed in particles of bacterium-like shape and dimension, the first two Acanthamoeba-infecting pandoraviruses remained up to now the most complex viruses since their discovery in 2013. Our isolation of three new strains from distant locations and environments is now used to perform the first comparative genomics analysis of the emerging worldwide-distributed Pandoraviridae family. Thorough annotation of the genomes combining transcriptomic, proteomic, and bioinformatic analyses reveals many non-coding transcripts and significantly reduces the former set of predicted protein-coding genes. Here we show that the pandoraviruses exhibit an open pan-genome, the enormous size of which is not adequately explained by gene duplications or horizontal transfers. As most of the strain-specific genes have no extant homolog and exhibit statistical features comparable to intergenic regions, we suggest that de novo gene creation could contribute to the evolution of the giant pandoravirus genomes.
随着 DNA 基因组达到 2.5Mb,并被包装成类似细菌形状和大小的颗粒,自 2013 年发现以来,前两种感染棘阿米巴的潘多拉病毒仍然是最复杂的病毒。我们从遥远的地点和环境中分离出三种新的毒株,现在用于对新兴的全球分布的潘多拉病毒科家族进行首次比较基因组分析。对基因组进行全面注释,结合转录组、蛋白质组和生物信息学分析,揭示了许多非编码转录本,并显著减少了以前预测的蛋白质编码基因。在这里,我们表明潘多拉病毒表现出开放的泛基因组,其巨大的大小不能仅通过基因重复或水平转移来解释。由于大多数菌株特异性基因没有现存的同源物,并表现出与基因间区可比的统计特征,我们认为从头基因的创造可能有助于巨型潘多拉病毒基因组的进化。