Cluster of Excellence on Multimodal Computing and Interaction (MMCI) and Max Planck Insitute for Informatics (MPII), Saarland University, Saarbrücken, Germany.
Saarbrücken Graduate School for Computer Science, Saarland University and International Max Planck Research School for Computer Science, Saarland Informatics Campus, Saarbrücken, Germany.
Sci Rep. 2019 Mar 26;9(1):5133. doi: 10.1038/s41598-019-41502-9.
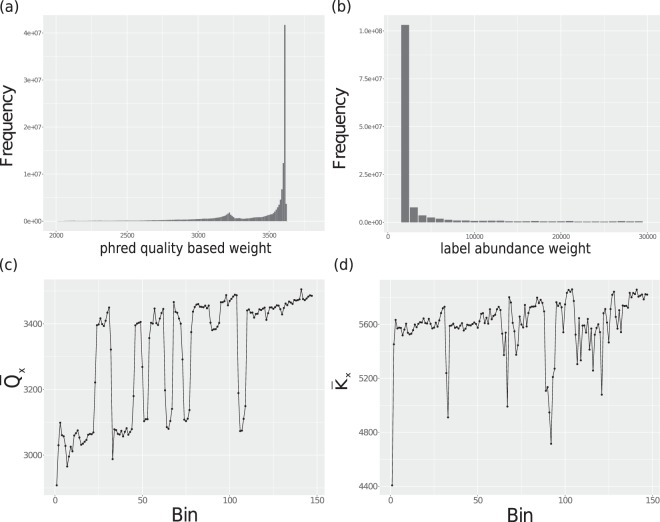
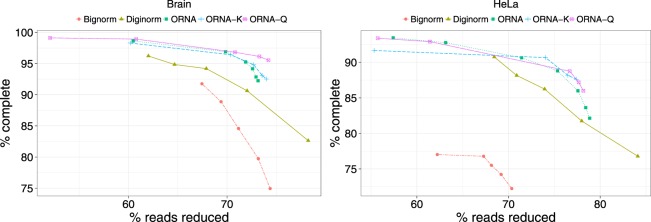
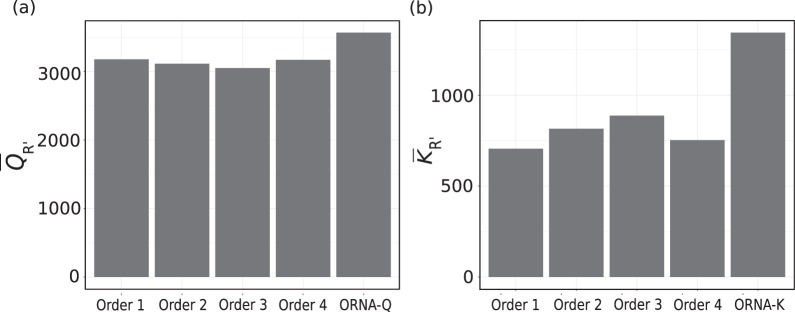
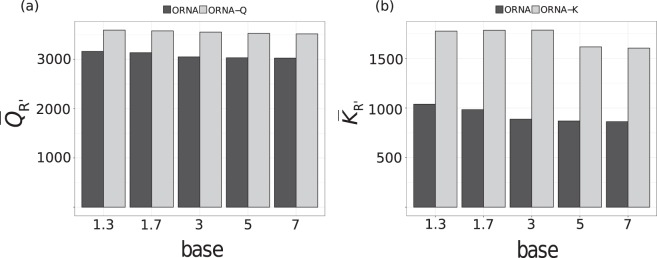
Specialized de novo assemblers for diverse datatypes have been developed and are in widespread use for the analyses of single-cell genomics, metagenomics and RNA-seq data. However, assembly of large sequencing datasets produced by modern technologies is challenging and computationally intensive. In-silico read normalization has been suggested as a computational strategy to reduce redundancy in read datasets, which leads to significant speedups and memory savings of assembly pipelines. Previously, we presented a set multi-cover optimization based approach, ORNA, where reads are reduced without losing important k-mer connectivity information, as used in assembly graphs. Here we propose extensions to ORNA, named ORNA-Q and ORNA-K, which consider a weighted set multi-cover optimization formulation for the in-silico read normalization problem. These novel formulations make use of the base quality scores obtained from sequencers (ORNA-Q) or k-mer abundances of reads (ORNA-K) to improve normalization further. We devise efficient heuristic algorithms for solving both formulations. In applications to human RNA-seq data, ORNA-Q and ORNA-K are shown to assemble more or equally many full length transcripts compared to other normalization methods at similar or higher read reduction values. The algorithm is implemented under the latest version of ORNA (v2.0, https://github.com/SchulzLab/ORNA ).
针对不同数据类型,已经开发出了专门的从头组装程序,并且广泛应用于单细胞基因组学、宏基因组学和 RNA-seq 数据分析。然而,利用现代技术生成的大型测序数据集的组装具有挑战性,并且计算密集度高。在计算中,reads 的数据归一化被认为是一种减少重复数据的计算策略,这可以显著提高组装流水线的速度和内存节省。在此之前,我们提出了一种基于多覆盖优化的方法 ORNA,该方法在不丢失重要的 k-mer 连接信息的情况下减少 reads 的数量,这些信息用于组装图。在这里,我们提出了 ORNA 的扩展,分别命名为 ORNA-Q 和 ORNA-K,它们考虑了用于在计算中进行 reads 归一化问题的加权多覆盖优化公式。这些新的公式利用从测序仪获得的碱基质量得分(ORNA-Q)或 reads 的 k-mer 丰度(ORNA-K)来进一步改进归一化。我们为这两种公式设计了高效的启发式算法来求解。在对人类 RNA-seq 数据的应用中,与其他归一化方法相比,ORNA-Q 和 ORNA-K 在相似或更高的 read 减少值下组装了更多或同等数量的全长转录本。该算法在最新版本的 ORNA(v2.0,https://github.com/SchulzLab/ORNA)下实现。