Cluster of Excellence on Multimodal Computing and Interaction, Saarland University, Saarbrücken, Germany.
Department of Computational Biology and Applied Algorithmics, Max Planck Institute for Informatics, Saarbrücken, Germany.
Bioinformatics. 2018 Oct 1;34(19):3273-3280. doi: 10.1093/bioinformatics/bty307.
De Bruijn graphs are a common assembly data structure for sequencing datasets. But with the advances in sequencing technologies, assembling high coverage datasets has become a computational challenge. Read normalization, which removes redundancy in datasets, is widely applied to reduce resource requirements. Current normalization algorithms, though efficient, provide no guarantee to preserve important k-mers that form connections between regions in the graph.
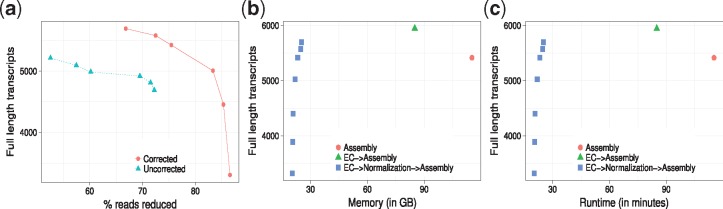
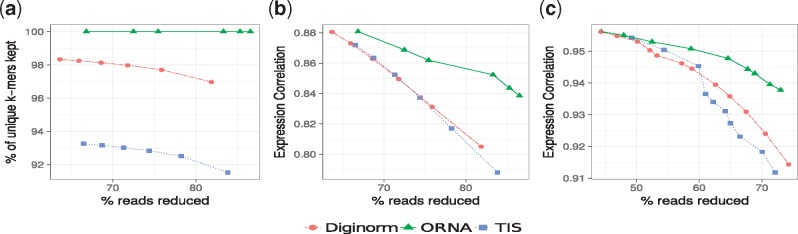
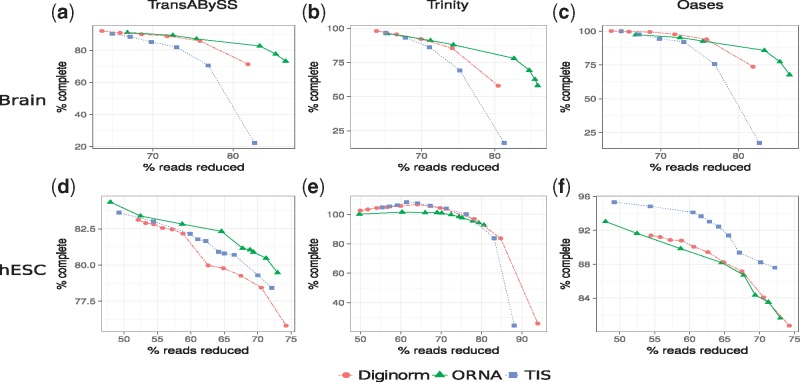
Here, normalization is phrased as a set multi-cover problem on reads and a heuristic algorithm, Optimized Read Normalization Algorithm (ORNA), is proposed. ORNA normalizes to the minimum number of reads required to retain all k-mers and their relative k-mer abundances from the original dataset. Hence, all connections from the original graph are preserved. ORNA was tested on various RNA-seq datasets with different coverage values. It was compared to the current normalization algorithms and was found to be performing better. Normalizing error corrected data allows for more accurate assemblies compared to the normalized uncorrected dataset. Further, an application is proposed in which multiple datasets are combined and normalized to predict novel transcripts that would have been missed otherwise. Finally, ORNA is a general purpose normalization algorithm that is fast and significantly reduces datasets with loss of assembly quality in between [1, 30]% depending on reduction stringency.
ORNA is available at https://github.com/SchulzLab/ORNA.
Supplementary data are available at Bioinformatics online.
De Bruijn 图是测序数据集的常用组装数据结构。但是,随着测序技术的进步,组装高覆盖率数据集已成为一个计算挑战。读段归一化(read normalization),用于去除数据集的冗余,被广泛应用于降低资源需求。虽然当前的归一化算法效率很高,但不能保证保留重要的 k-mer,这些 k-mer 构成了图中区域之间的连接。
在这里,归一化被表述为一个关于读段的集多重覆盖问题,并提出了一种启发式算法,即优化读段归一化算法(Optimized Read Normalization Algorithm,ORNA)。ORNA 将归一化到保留原始数据集的所有 k-mer 及其相对 k-mer 丰度所需的最少读段数。因此,保留了原始图的所有连接。ORNA 在具有不同覆盖值的各种 RNA-seq 数据集上进行了测试。与当前的归一化算法相比,它的性能更好。与归一化未纠错数据相比,对纠错后的数据进行归一化可以得到更准确的组装结果。此外,还提出了一个应用程序,它可以组合和归一化多个数据集,以预测可能会错过的新转录本。最后,ORNA 是一种通用的归一化算法,速度快,在 [1, 30]%的缩减严格性之间显著减少数据集,而不会损失组装质量。
ORNA 可在 https://github.com/SchulzLab/ORNA 上获得。
补充数据可在 Bioinformatics 在线获取。