1Parasites and Microbes, Wellcome Trust Sanger Institute, Wellcome Genome Campus, Hinxton CB10 1SA, UK.
2London School of Hygiene & Tropical Medicine, Keppel St., London WC1E 7HT, UK.
Microb Genom. 2019 Apr;5(4). doi: 10.1099/mgen.0.000264. Epub 2019 Mar 28.
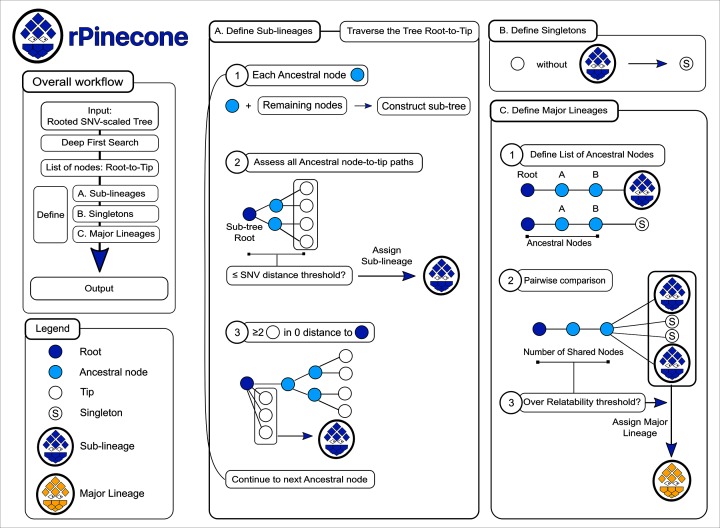
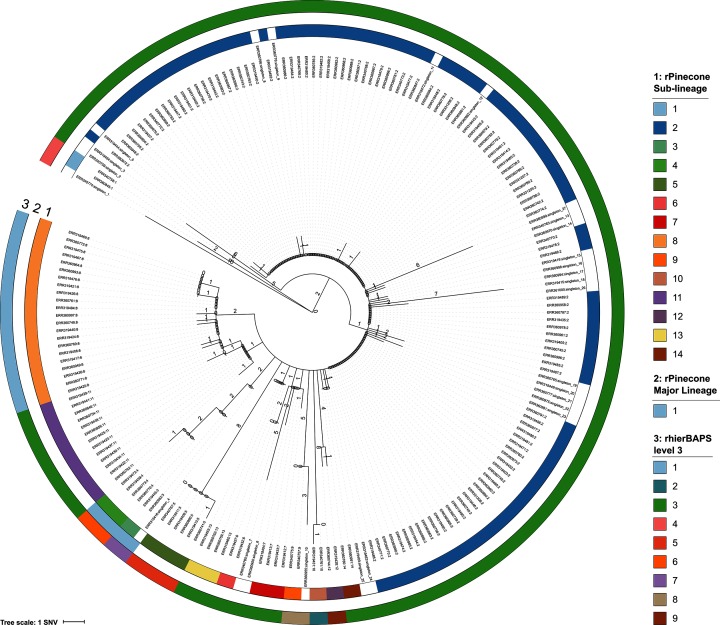
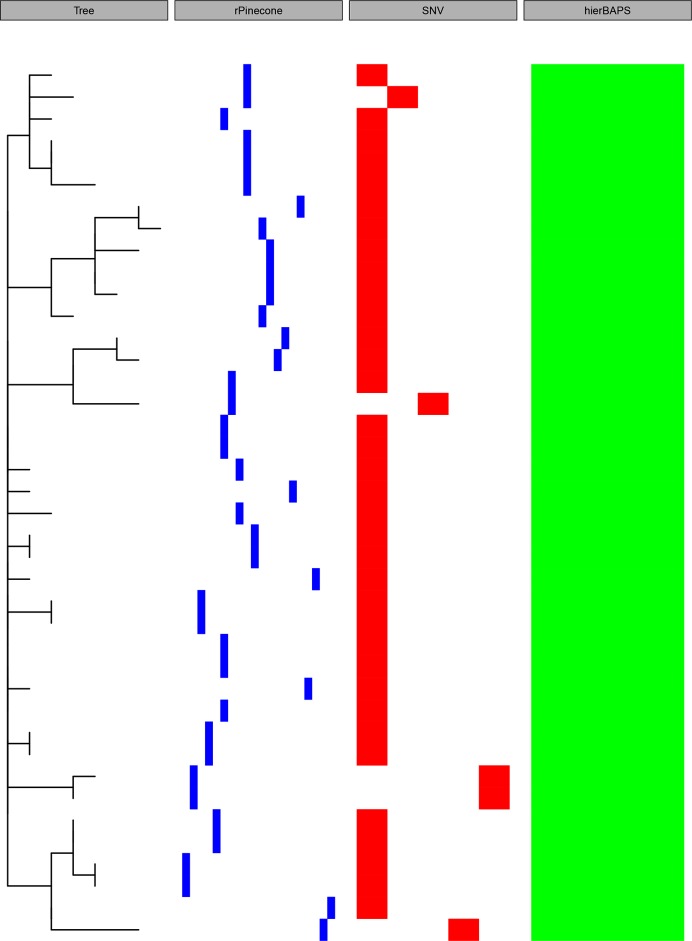
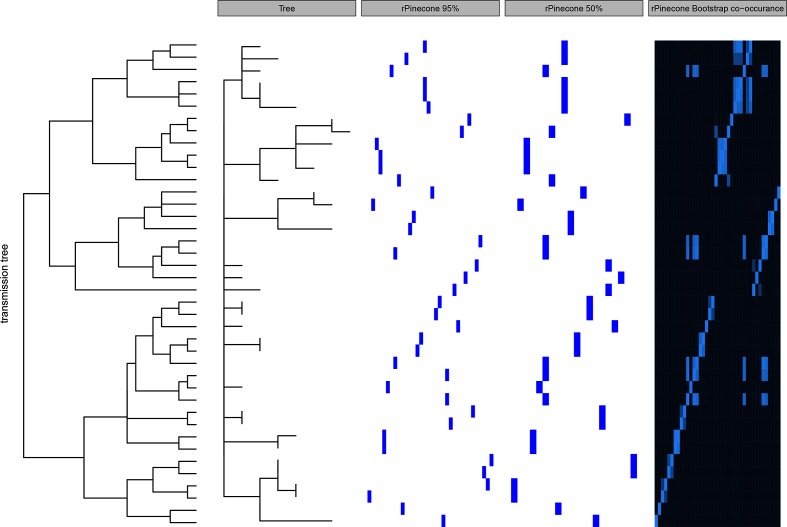
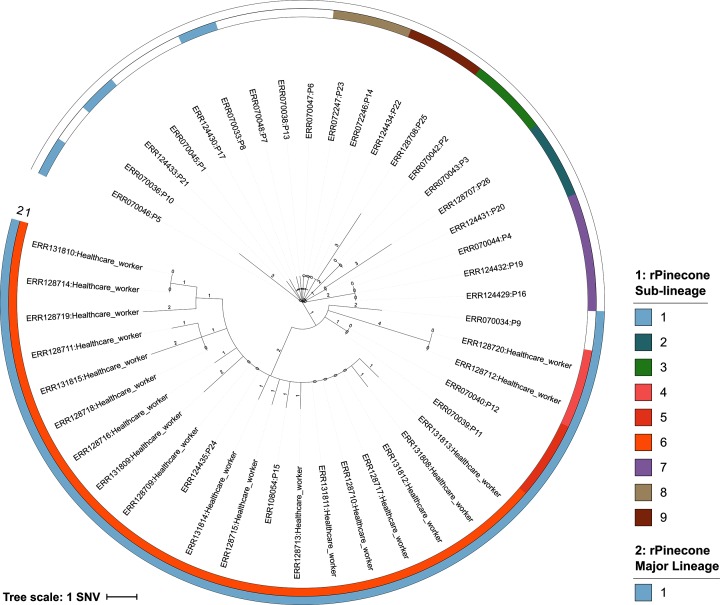
The ability to distinguish different circulating pathogen clones from each other is a fundamental requirement to understand the epidemiology of infectious diseases. Phylogenetic analysis of genomic data can provide a powerful platform to identify lineages within bacterial populations, and thus inform outbreak investigation and transmission dynamics. However, resolving differences between pathogens associated with low-variant (LV) populations carrying low median pairwise single nucleotide variant (SNV) distances remains a major challenge. Here we present rPinecone, an R package designed to define sub-lineages within closely related LV populations. rPinecone uses a root-to-tip directional approach to define sub-lineages within a phylogenetic tree according to SNV distance from the ancestral node. The utility of this software was demonstrated using both simulated outbreaks and real genomic data of two LV populations: a hospital outbreak of methicillin-resistant Staphylococcus aureus and endemic Salmonella Typhi from rural Cambodia. rPinecone identified the transmission branches of the hospital outbreak and geographically confined lineages in Cambodia. Sub-lineages identified by rPinecone in both analyses were phylogenetically robust. It is anticipated that rPinecone can be used to discriminate between lineages of bacteria from LV populations where other methods fail, enabling a deeper understanding of infectious disease epidemiology for public health purposes.
能够区分彼此之间不同的循环病原体克隆是理解传染病流行病学的基本要求。对基因组数据进行系统发育分析可以为识别细菌种群中的谱系提供一个强大的平台,从而为暴发调查和传播动力学提供信息。然而,解决与携带低中位数成对单核苷酸变异 (SNV) 距离的低变异 (LV) 种群相关的病原体之间的差异仍然是一个主要挑战。在这里,我们介绍了 rPinecone,这是一个专为定义密切相关的 LV 种群内的亚谱系而设计的 R 包。rPinecone 使用根到尖端的方向方法,根据与祖先节点的 SNV 距离在系统发育树中定义亚谱系。该软件的实用性通过模拟暴发和来自柬埔寨农村的两种 LV 人群的真实基因组数据得到了证明:耐甲氧西林金黄色葡萄球菌的医院暴发和地方性伤寒沙门氏菌。rPinecone 确定了医院暴发的传播分支和柬埔寨的地理限定谱系。rPinecone 在这两种分析中识别的亚谱系在系统发育上是稳健的。预计 rPinecone 可用于区分来自 LV 人群的细菌谱系,而其他方法则无法区分,从而为公共卫生目的深入了解传染病流行病学。