Kalafatovic Daniela, Mauša Goran, Todorovski Toni, Giralt Ernest
Institute for Research in Biomedicine (IRB Barcelona), The Barcelona Institute of Science and Technology (BIST), Baldiri Reixac 10, 08028, Barcelona, Spain.
Faculty of Engineering, University of Rijeka, Vukovarska 58, 51000, Rijeka, Croatia.
J Cheminform. 2019 Mar 28;11(1):25. doi: 10.1186/s13321-019-0347-6.
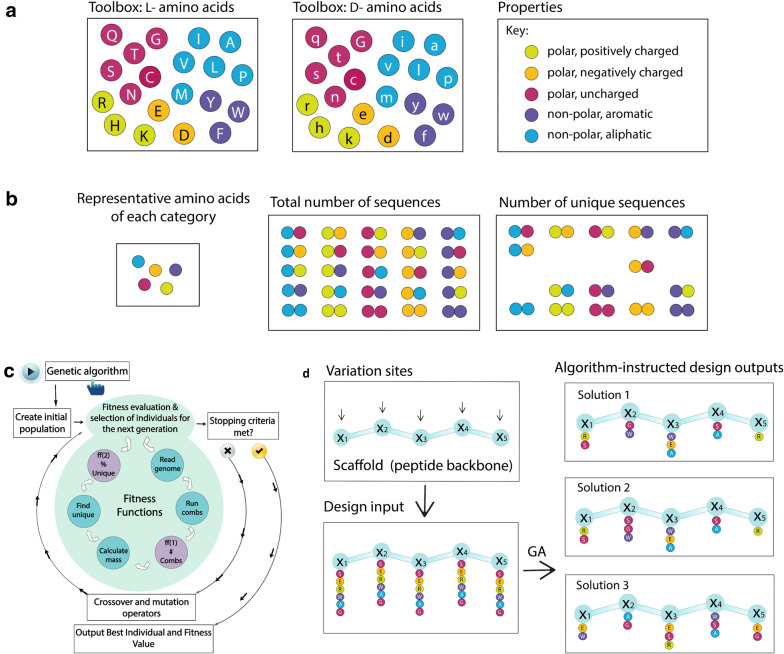
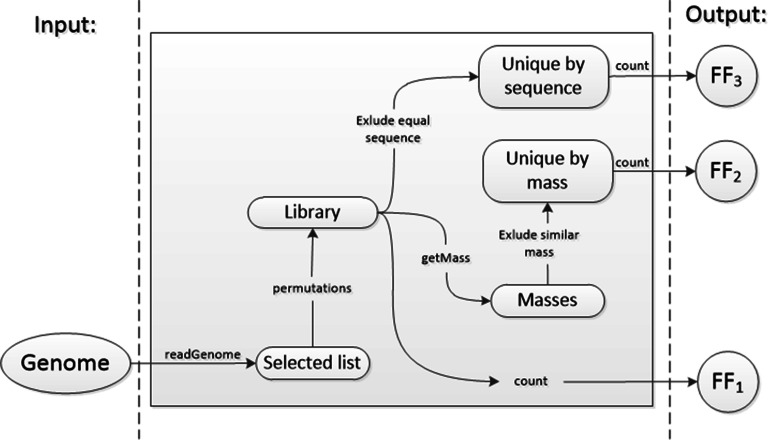
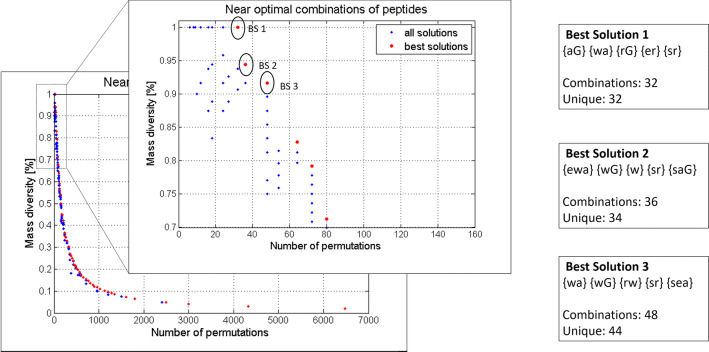
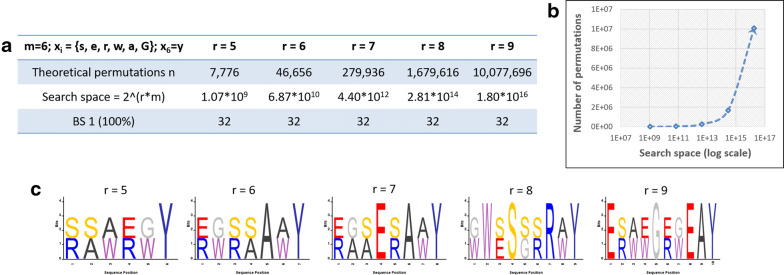
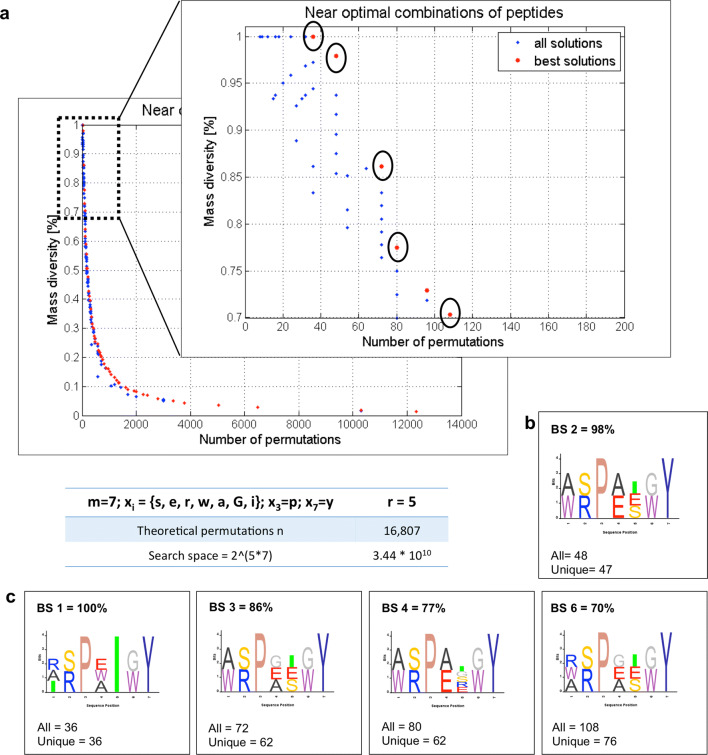
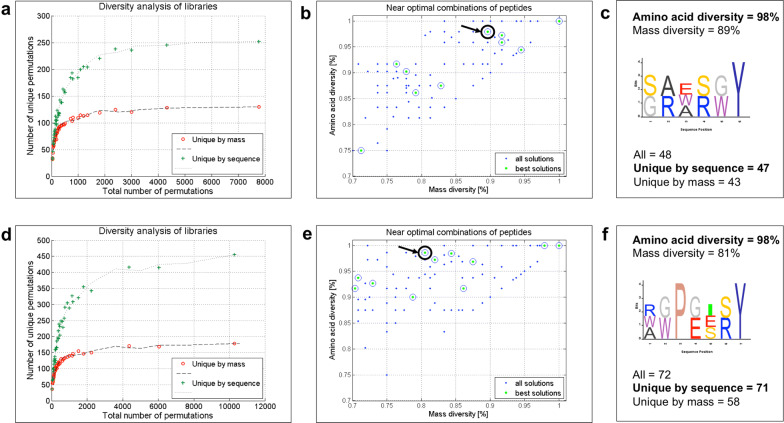
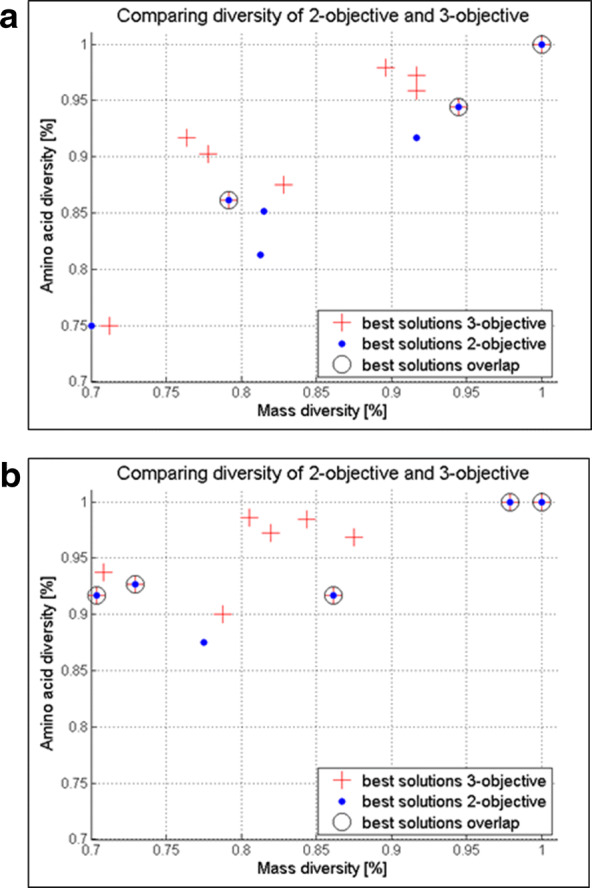
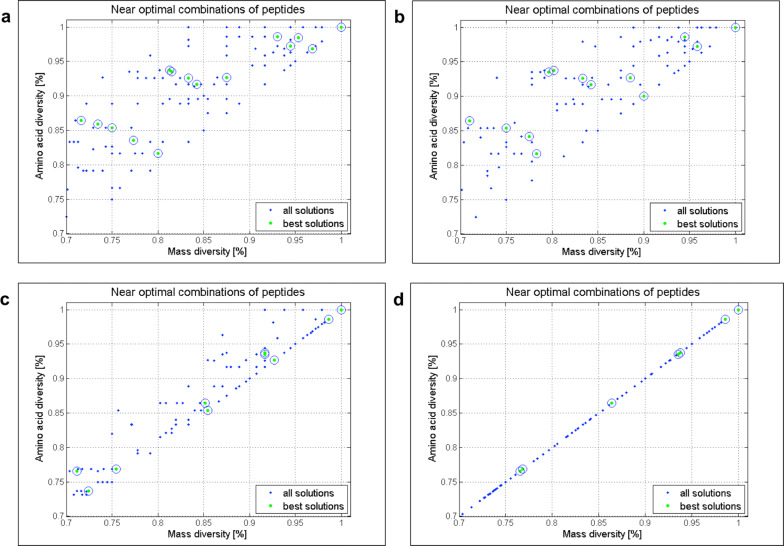
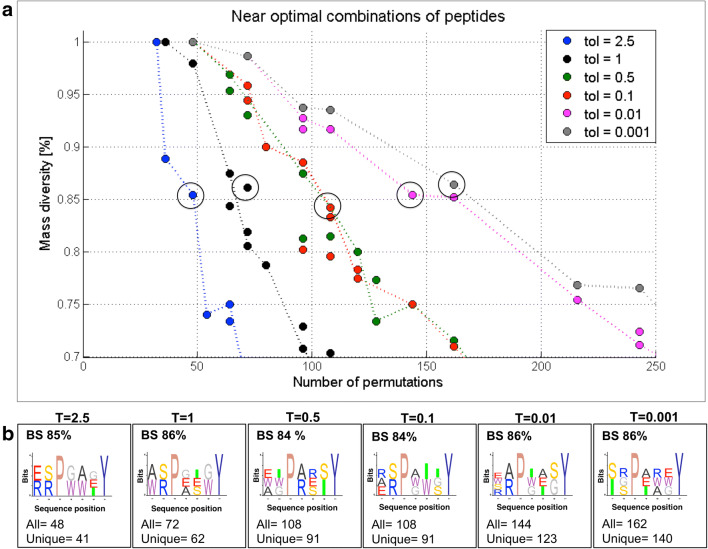
Random peptide libraries that cover large search spaces are often used for the discovery of new binders, even when the target is unknown. To ensure an accurate population representation, there is a tendency to use large libraries. However, parameters such as the synthesis scale, the number of library members, the sequence deconvolution and peptide structure elucidation, are challenging when increasing the library size. To tackle these challenges, we propose an algorithm-supported approach to peptide library design based on molecular mass and amino acid diversity. The aim is to simplify the tedious permutation identification in complex mixtures, when mass spectrometry is used, by avoiding mass redundancy. For this purpose, we applied multi (two- and three-)-objective genetic algorithms to discriminate between library members based on defined parameters. The optimizations led to diverse random libraries by maximizing the number of amino acid permutations and minimizing the mass and/or sequence overlapping. The algorithm-suggested designs offer to the user a choice of appropriate compromise solutions depending on the experimental needs. This implies that diversity rather than library size is the key element when designing peptide libraries for the discovery of potential novel biologically active peptides.
即使目标未知,覆盖大搜索空间的随机肽库也常用于发现新的结合物。为确保准确的群体代表性,人们倾向于使用大型文库。然而,当增加文库规模时,诸如合成规模、文库成员数量、序列去卷积和肽结构解析等参数具有挑战性。为应对这些挑战,我们提出一种基于分子量和氨基酸多样性的算法支持的肽库设计方法。目的是在使用质谱时,通过避免质量冗余来简化复杂混合物中繁琐的排列鉴定。为此,我们应用多(二和三)目标遗传算法,根据定义的参数区分文库成员。通过最大化氨基酸排列数量并最小化质量和/或序列重叠,优化产生了多样化的随机文库。算法建议的设计根据实验需求为用户提供了合适的折衷解决方案选择。这意味着在设计用于发现潜在新型生物活性肽的肽库时,多样性而非文库规模是关键要素。