Grupo de Investigación Bioinformática y Genómica Funcional. Laboratorio 19. Centro de Investigación del Cáncer (CiC-IBMCC, Universidad de Salamanca-CSIC, Campus Universitario Miguel de Unamuno s/n, Salamanca, 37007, Spain.
Universidad de La Frontera. Centro De Excelencia de Modelación y Computación Científica, C/ Montevideo 740, Temuco, Chile.
BMC Genomics. 2019 Apr 2;20(1):259. doi: 10.1186/s12864-019-5496-5.
RNA sequencing is a widely used technology for differential expression analysis. However, the RNA-Seq do not provide accurate absolute measurements and the results can be different for each pipeline used. The major problem in statistical analysis of RNA-Seq and in the omics data in general, is the small sample size with respect to the large number of variables. In addition, experimental design must be taken into account and few tools consider it.
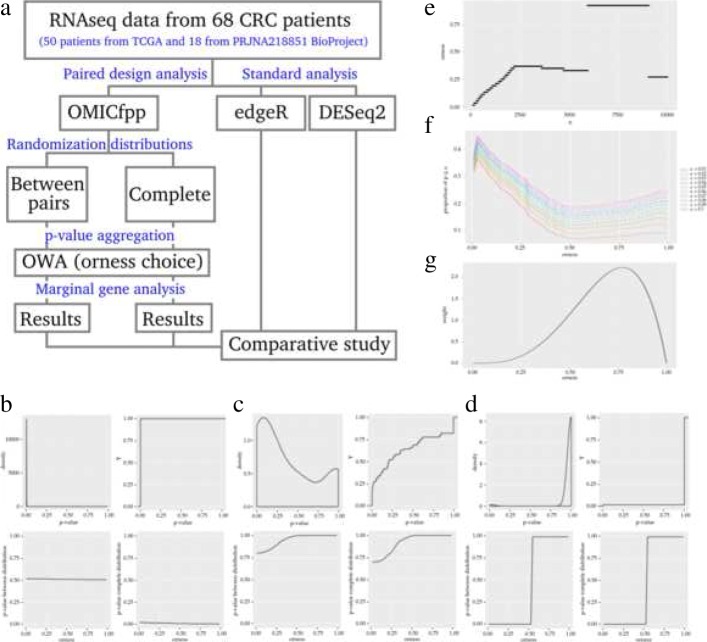
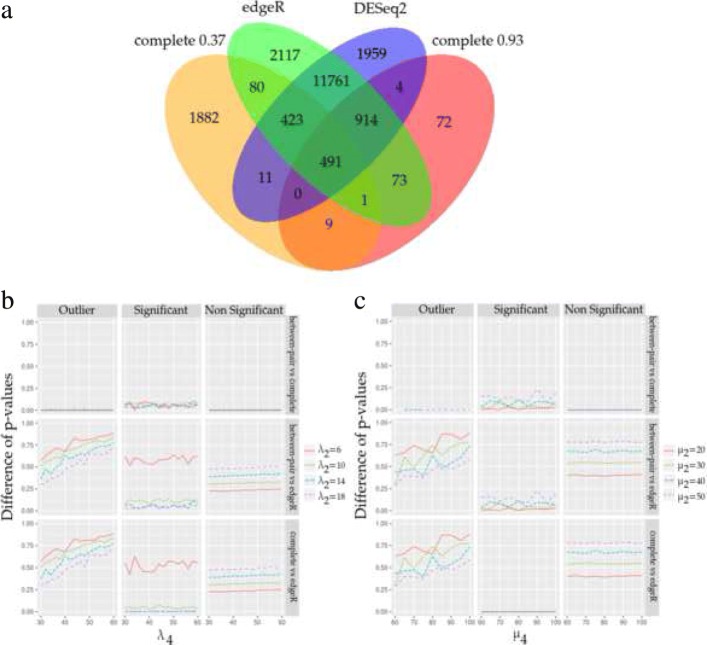
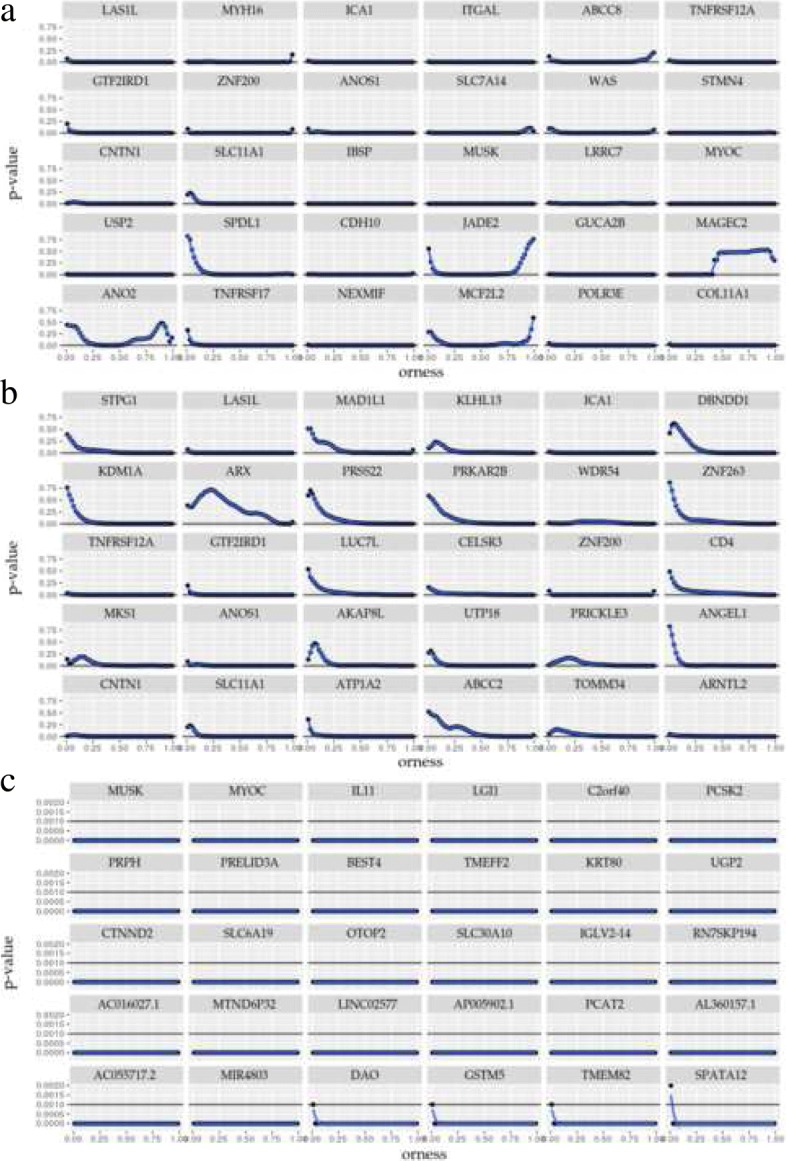
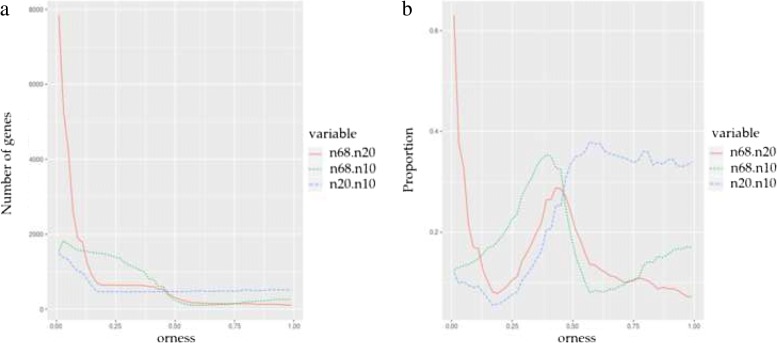
We propose OMICfpp, a method for the statistical analysis of RNA-Seq paired design data. First, we obtain a p-value for each case-control pair using a binomial test. These p-values are aggregated using an ordered weighted average (OWA) with a given orness previously chosen. The aggregated p-value from the original data is compared with the aggregated p-value obtained using the same method applied to random pairs. These new pairs are generated using between-pairs and complete randomization distributions. This randomization p-value is used as a raw p-value to test the differential expression of each gene. The OMICfpp method is evaluated using public data sets of 68 sample pairs from patients with colorectal cancer. We validate our results through bibliographic search of the reported genes and using simulated data set. Furthermore, we compared our results with those obtained by the methods edgeR and DESeq2 for paired samples. Finally, we propose new target genes to validate these as gene expression signatures in colorectal cancer. OMICfpp is available at http://www.uv.es/ayala/software/OMICfpp_0.2.tar.gz .
Our study shows that OMICfpp is an accurate method for differential expression analysis in RNA-Seq data with paired design. In addition, we propose the use of randomized p-values pattern graphic as a powerful and robust method to select the target genes for experimental validation.
RNA 测序是一种广泛用于差异表达分析的技术。然而,RNA-Seq 不能提供准确的绝对测量值,并且每个使用的管道的结果可能不同。在 RNA-Seq 的统计分析以及一般的组学数据中,主要问题是相对于大量变量而言,样本量较小。此外,必须考虑实验设计,而很少有工具考虑到这一点。
我们提出了 OMICfpp,这是一种用于 RNA-Seq 配对设计数据的统计分析方法。首先,我们使用二项式检验为每个病例对照对获得一个 p 值。这些 p 值使用给定的有序权重平均 (OWA) 进行聚合,该平均权重是之前选择的。原始数据的聚合 p 值与使用相同方法应用于随机对获得的聚合 p 值进行比较。这些新对是使用对间和完全随机化分布生成的。将该随机化 p 值用作测试每个基因差异表达的原始 p 值。使用来自结直肠癌患者的 68 个样本对的公共数据集评估 OMICfpp 方法。我们通过报告基因的文献搜索和使用模拟数据集验证了我们的结果。此外,我们将我们的结果与配对样本的 edgeR 和 DESeq2 方法的结果进行了比较。最后,我们提出了新的靶基因来验证这些作为结直肠癌的基因表达特征。OMICfpp 可在 http://www.uv.es/ayala/software/OMICfpp_0.2.tar.gz 获得。
我们的研究表明,OMICfpp 是一种用于 RNA-Seq 数据中配对设计的差异表达分析的准确方法。此外,我们建议使用随机化 p 值模式图作为选择用于实验验证的靶基因的强大而稳健的方法。