Department of Biology, Faculty of Medicine and Dentistry, Palacky University, Olomouc, Czech Republic.
Department of Histology and Embryology, Faculty of Medicine and Dentistry, Palacky University, Olomouc, Czech Republic.
Oncogene. 2019 Jul;38(28):5627-5642. doi: 10.1038/s41388-019-0813-7. Epub 2019 Apr 9.
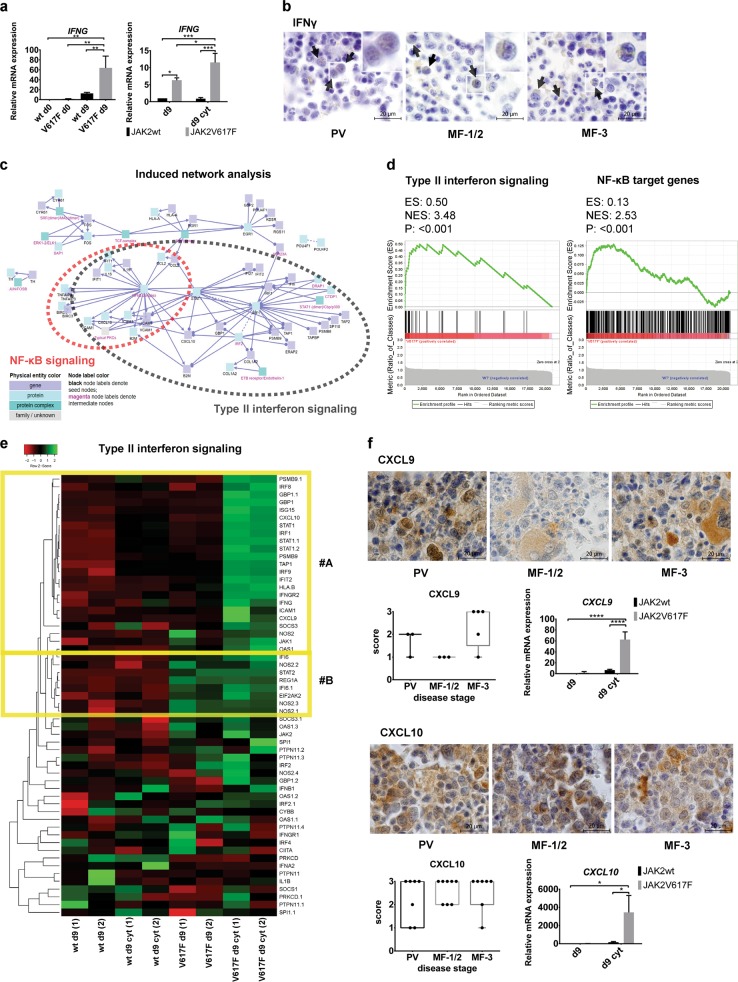
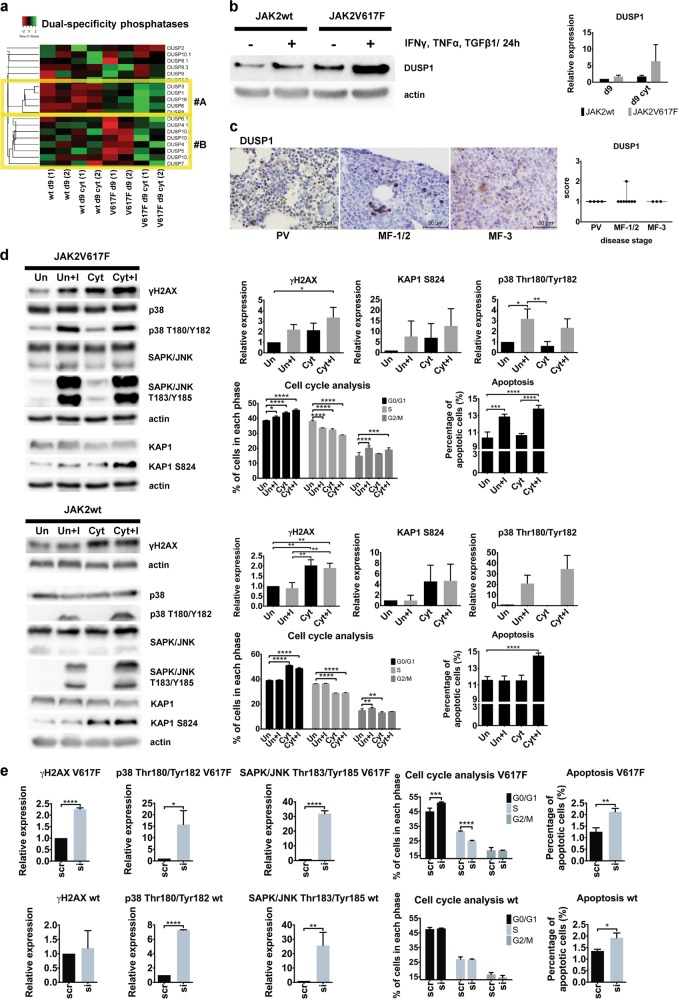
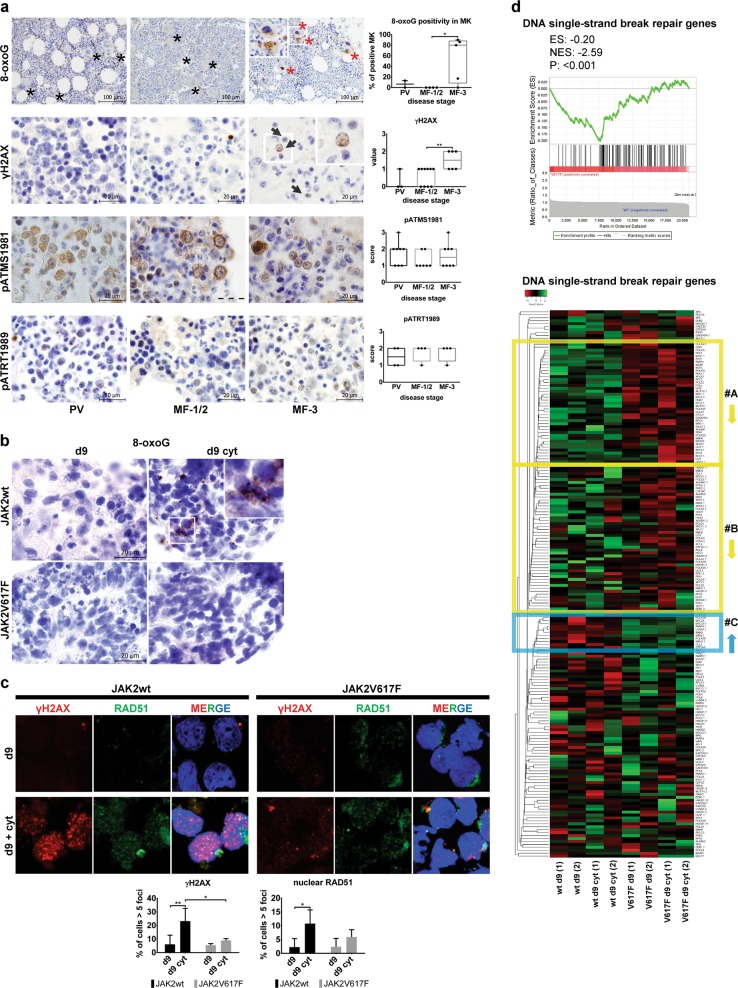
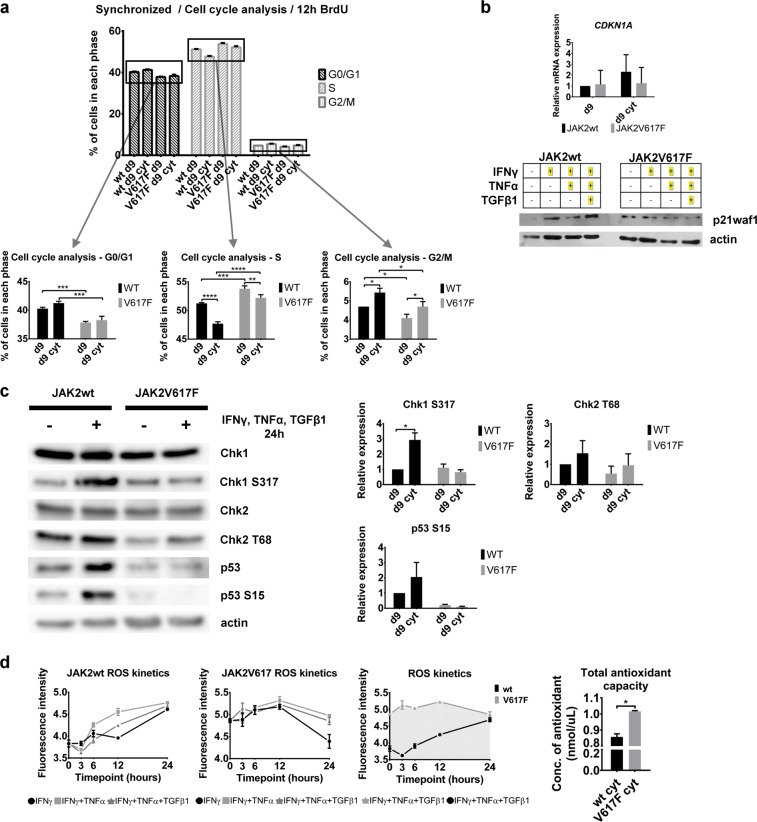
Inflammatory and oncogenic signaling converge in disease evolution of BCR-ABL-negative myeloproliferative neoplasms, clonal hematopoietic stem cell disorders characterized by gain-of-function mutation in JAK2 kinase (JAK2V617F), with highest prevalence in patients with polycythemia vera (PV). Despite the high risk, DNA-damaging inflammatory microenvironment, PV progenitors tend to preserve their genomic stability over decades until their progression to post-PV myelofibrosis/acute myeloid leukemia. Using induced pluripotent stem cells-derived CD34 progenitor-enriched cultures from JAK2V617F PV patient and from JAK2 wild-type healthy control, CRISPR-modified HEL cells and patients' bone marrow sections from different disease stages, we demonstrate that JAK2V617F induces an intrinsic IFNγ- and NF-κB-associated inflammatory program, while suppressing inflammation-evoked DNA damage both in vitro and in vivo. We show that cells with JAK2V617F tightly regulate levels of inflammatory cytokines-induced reactive oxygen species, do not fully activate the ATM/p53/p21waf1 checkpoint and p38/JNK MAPK stress pathway signaling when exposed to inflammatory cytokines, suppress DNA single-strand break repair genes' expression yet overexpress the dual-specificity phosphatase (DUSP) 1. RNAi-mediated knock-down and pharmacological inhibition of DUSP1, involved in p38/JNK deactivation, in HEL cells reveals growth addiction to DUSP1, consistent with enhanced DNA damage response and apoptosis in DUSP1-inhibited parental JAK2V617F cells, but not in CRISPR-modified JAK2 wild-type cells. Our results indicate that the JAK2V617F PV progenitors utilize DUSP1 activity as a protection mechanism against DNA damage accumulation, promoting their proliferation and survival in the inflammatory microenvironment, identifying DUSP1 as a potential therapeutic target in PV.
在 BCR-ABL 阴性骨髓增殖性肿瘤(MPN)的疾病演变过程中,炎症和致癌信号汇聚在一起,这些疾病是由 JAK2 激酶(JAK2V617F)功能获得性突变引起的克隆性造血干细胞疾病,其中以真性红细胞增多症(PV)患者中最为常见。尽管存在高风险的 DNA 损伤炎症微环境,但 PV 祖细胞往往能在几十年内保持其基因组稳定性,直到发展为 PV 后骨髓纤维化/急性髓系白血病。我们使用 JAK2V617F PV 患者和 JAK2 野生型健康对照的诱导多能干细胞(iPSC)衍生的 CD34 祖细胞富集培养物、CRISPR 修饰的 HEL 细胞和不同疾病阶段的患者骨髓切片,证明 JAK2V617F 诱导内在的 IFNγ 和 NF-κB 相关炎症程序,同时抑制体外和体内炎症诱导的 DNA 损伤。我们发现 JAK2V617F 细胞严格调控由炎症细胞因子诱导的活性氧水平,当暴露于炎症细胞因子时,不完全激活 ATM/p53/p21waf1 检查点和 p38/JNK MAPK 应激途径信号,抑制 DNA 单链断裂修复基因的表达,但过度表达双特异性磷酸酶(DUSP)1。在 HEL 细胞中,通过 RNAi 介导的敲低和 DUSP1 的药理学抑制,该酶参与 p38/JNK 的失活,揭示了 HEL 细胞对 DUSP1 的生长依赖,这与 DUSP1 抑制的 JAK2V617F 亲本细胞中增强的 DNA 损伤反应和细胞凋亡一致,但在 CRISPR 修饰的 JAK2 野生型细胞中则不然。我们的结果表明,JAK2V617F PV 祖细胞利用 DUSP1 活性作为一种防止 DNA 损伤积累的保护机制,促进其在炎症微环境中的增殖和存活,确定 DUSP1 为 PV 的潜在治疗靶点。