Department of Ophthalmic Research, Cole Eye Institute, Cleveland Clinic, Cleveland, Ohio, United States of America.
Department of Molecular Medicine, Cleveland Clinic Lerner College of Medicine, Case Western Reserve University, Cleveland, Ohio, United States of America.
PLoS One. 2019 Apr 10;14(4):e0213960. doi: 10.1371/journal.pone.0213960. eCollection 2019.
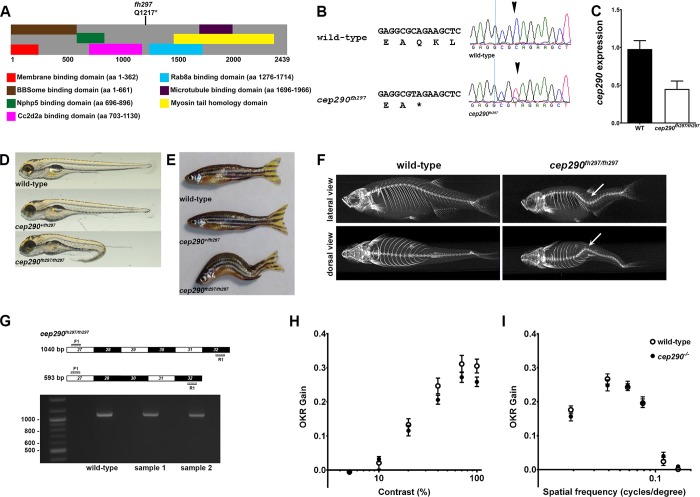
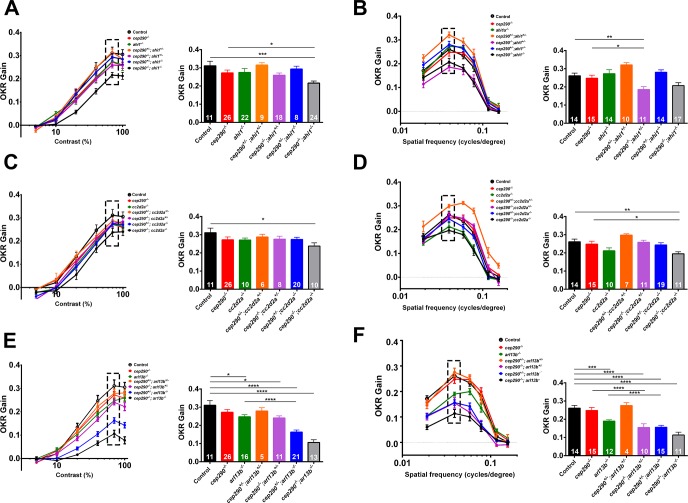
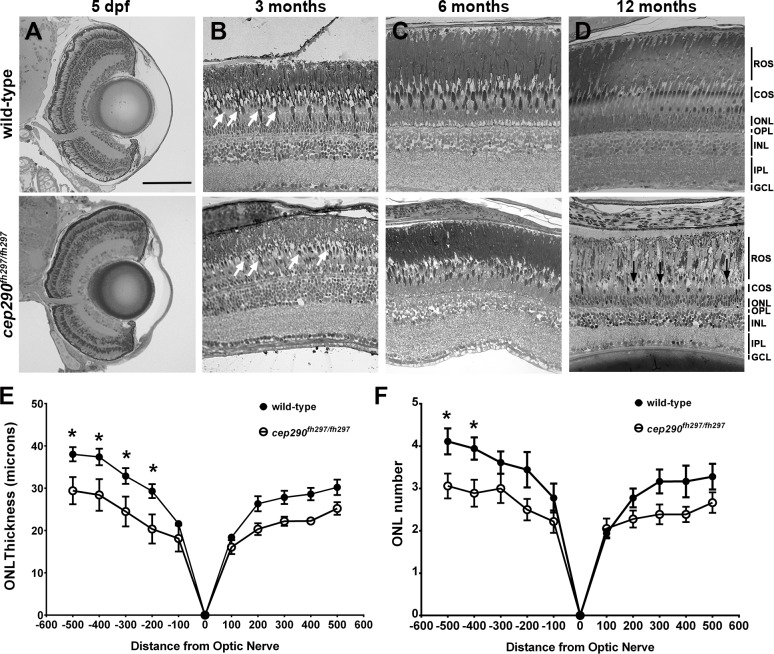
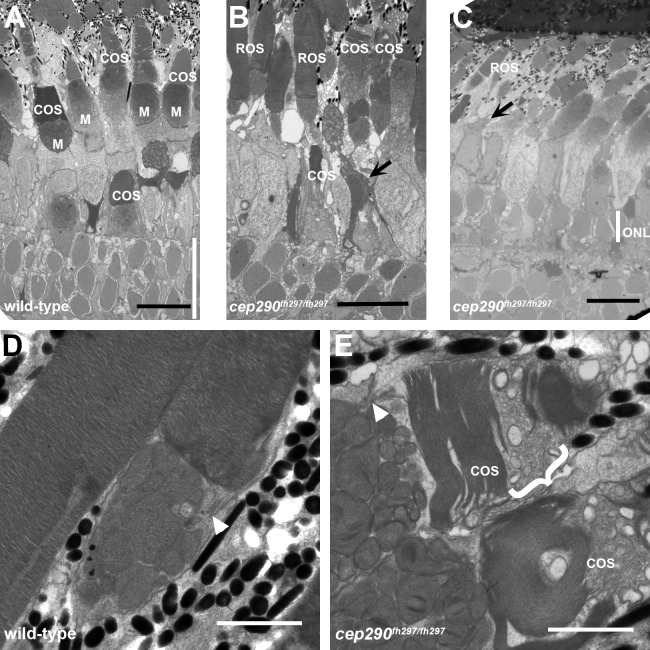
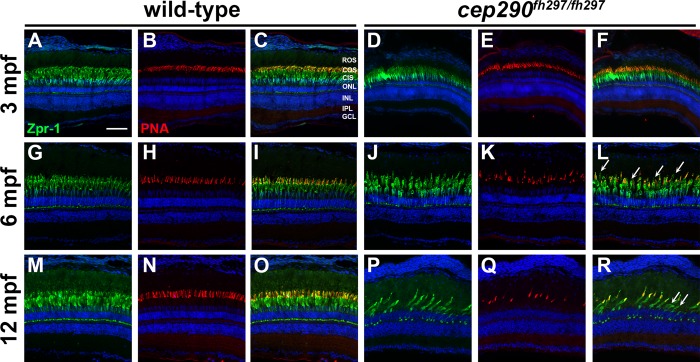
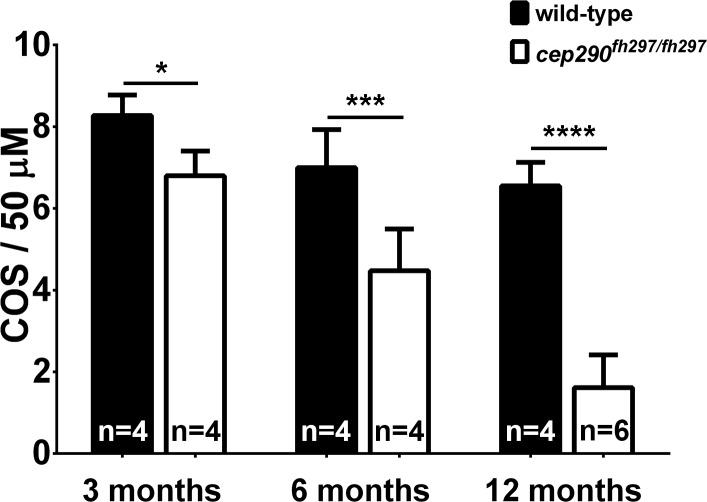
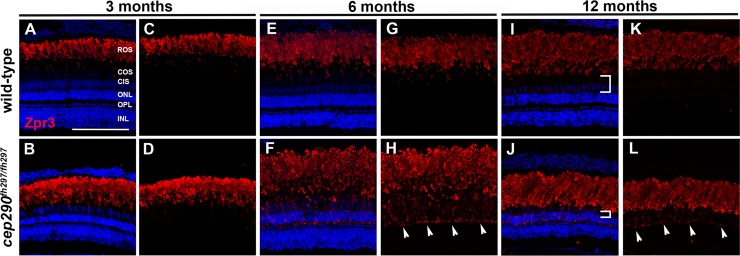
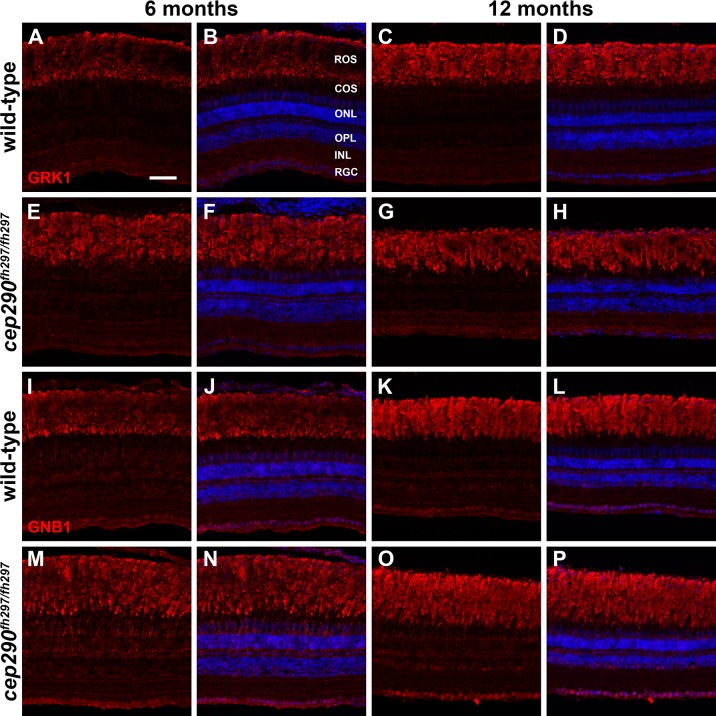
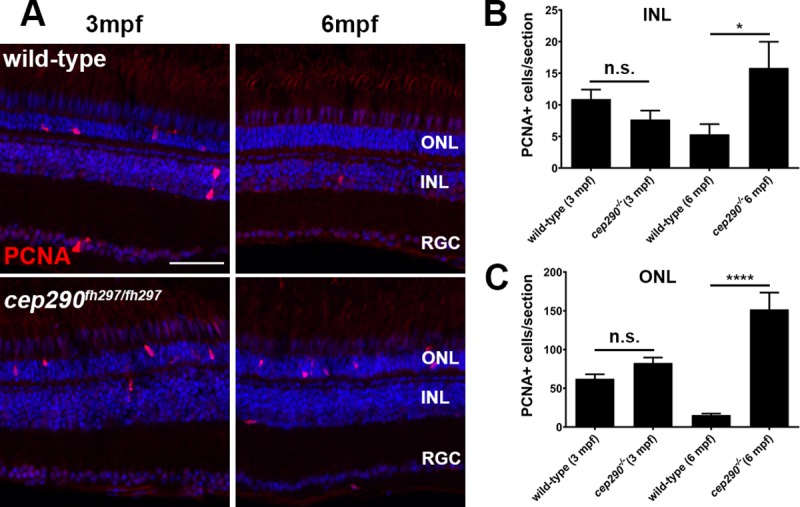
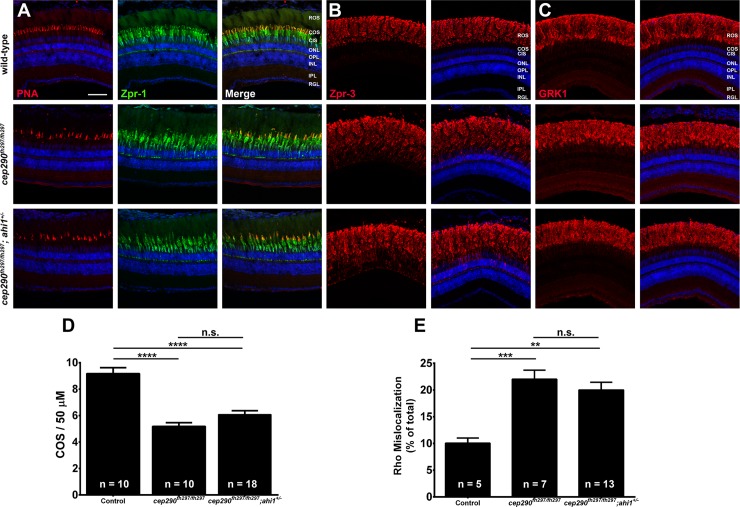
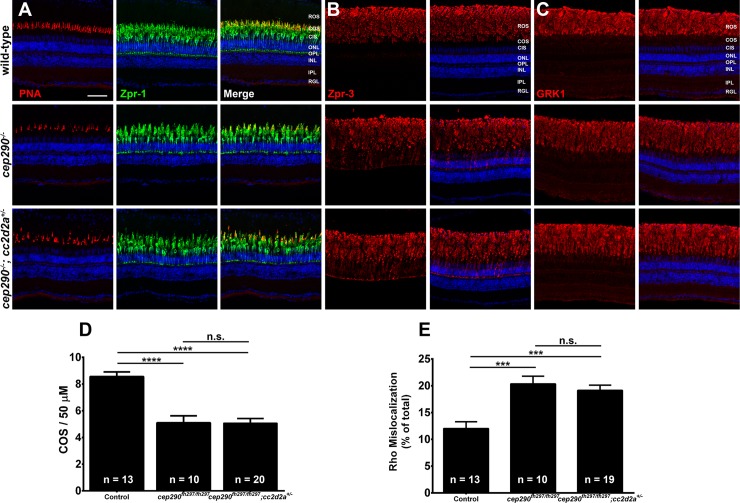
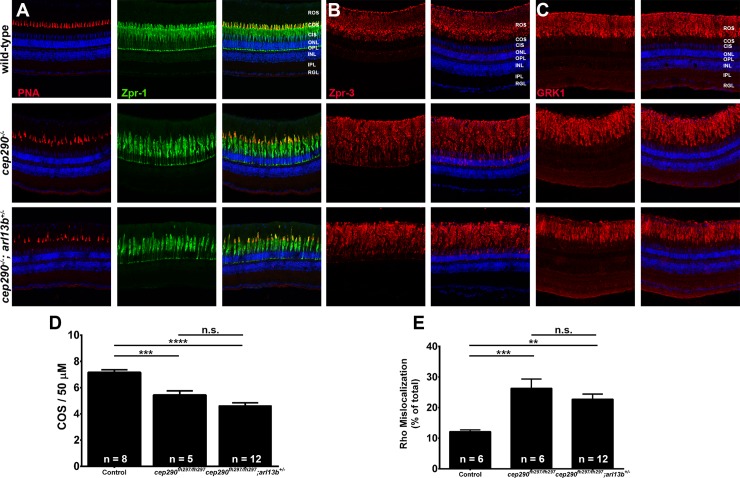
Mutations in the gene Centrosomal Protein 290 kDa (CEP290) result in multiple ciliopathies ranging from the neonatal lethal disorder Meckel-Gruber Syndrome to multi-systemic disorders such as Joubert Syndrome and Bardet-Biedl Syndrome to nonsyndromic diseases like Leber Congenital Amaurosis (LCA) and retinitis pigmentosa. Results from model organisms and human genetics studies, have suggest that mutations in genes encoding protein components of the transition zone (TZ) and other cilia-associated proteins can function as genetic modifiers and be a source for CEP290 pleiotropy. We investigated the zebrafish cep290fh297/fh297 mutant, which encodes a nonsense mutation (p.Q1217*). This mutant is viable as adults, exhibits scoliosis, and undergoes a slow, progressive cone degeneration. The cep290fh297/fh297 mutants showed partial mislocalization of the transmembrane protein rhodopsin but not of the prenylated proteins rhodopsin kinase (GRK1) or the rod transducin subunit GNB1. Surprisingly, photoreceptor degeneration did not trigger proliferation of Müller glia, but proliferation of rod progenitors in the outer nuclear layer was significantly increased. To determine if heterozygous mutations in other cilia genes could exacerbate retinal degeneration, we bred cep290fh297/fh297 mutants to arl13b, ahi1, and cc2d2a mutant zebrafish lines. While cep290fh297/fh297 mutants lacking a single allele of these genes did not exhibit accelerated photoreceptor degeneration, loss of one alleles of arl13b or ahi1 reduced visual performance in optokinetic response assays at 5 days post fertilization. Our results indicate that the cep290fh297/fh297 mutant is a useful model to study the role of genetic modifiers on photoreceptor degeneration in zebrafish and to explore how progressive photoreceptor degeneration influences regeneration in adult zebrafish.
CEP290 基因突变导致多种纤毛病,从新生儿致死性疾病梅克尔-格鲁伯综合征(Meckel-Gruber Syndrome)到多系统疾病,如杰伯综合征(Joubert Syndrome)和巴德-比德尔综合征(Bardet-Biedl Syndrome),再到非综合征性疾病,如莱伯先天性黑蒙(Leber Congenital Amaurosis,LCA)和色素性视网膜炎。来自模式生物和人类遗传学研究的结果表明,编码过渡区(transition zone,TZ)和其他纤毛相关蛋白的蛋白成分的基因突变可以作为遗传修饰因子,并成为 CEP290 多效性的来源。我们研究了斑马鱼 cep290fh297/fh297 突变体,该突变体编码一个无义突变(p.Q1217*)。该突变体成体存活,表现为脊柱侧凸,并经历缓慢、进行性的锥体变性。cep290fh297/fh297 突变体显示跨膜蛋白视蛋白的部分定位错误,但prenylated 蛋白视蛋白激酶(GRK1)或 rod 转导亚基 GNB1 没有定位错误。令人惊讶的是,光感受器变性并没有触发 Muller 胶质细胞的增殖,但外核层中的 rod 前体细胞增殖显著增加。为了确定其他纤毛病基因的杂合突变是否会加剧视网膜变性,我们将 cep290fh297/fh297 突变体与 arl13b、ahi1 和 cc2d2a 突变体斑马鱼系杂交。虽然这些基因的单个等位基因缺失的 cep290fh297/fh297 突变体没有表现出加速的光感受器变性,但 arl13b 或 ahi1 的一个等位基因缺失降低了 5 天龄受精后在视动反应测试中的视觉表现。我们的结果表明,cep290fh297/fh297 突变体是研究遗传修饰因子对斑马鱼光感受器变性作用的有用模型,并探索进行性光感受器变性如何影响成年斑马鱼的再生。