Department of Microbiology & Plant Pathology and Institute for Integrative Genome Biology, University of California-Riverside, Riverside, CA, 92521, USA.
Present address: Critical Care Medicine Department, NIH Clinical Center, National Institutes of Health, Bethesda, MD, USA.
BMC Bioinformatics. 2019 Apr 15;20(1):184. doi: 10.1186/s12859-019-2782-9.
Inexpensive high-throughput DNA sequencing has democratized access to genetic information for most organisms so that research utilizing a genome or transcriptome of an organism is not limited to model systems. However, the quality of the assemblies of sampled genomes can vary greatly which hampers utility for comparisons and meaningful interpretation. The uncertainty of the completeness of a given genome sequence can limit feasibility of asserting patterns of high rates of gene loss reported in many lineages.
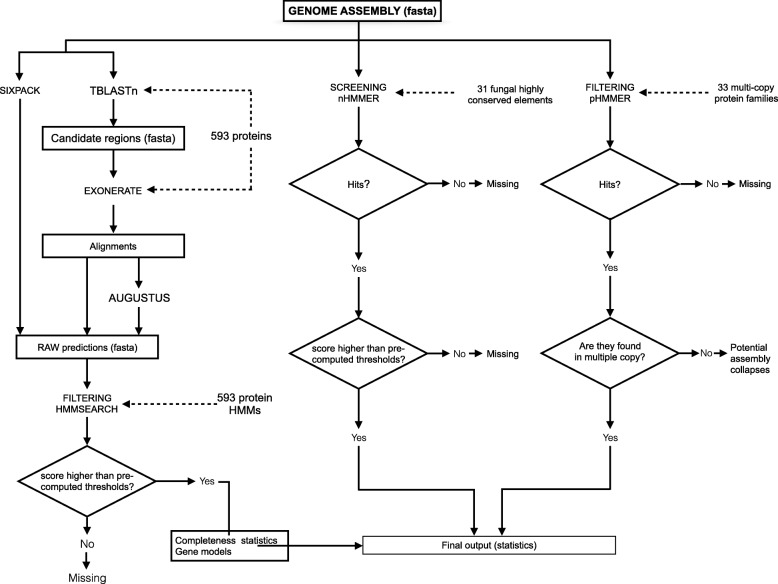
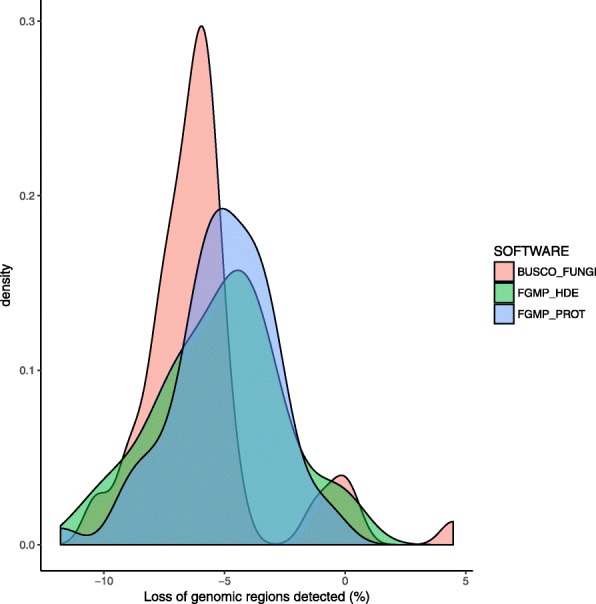
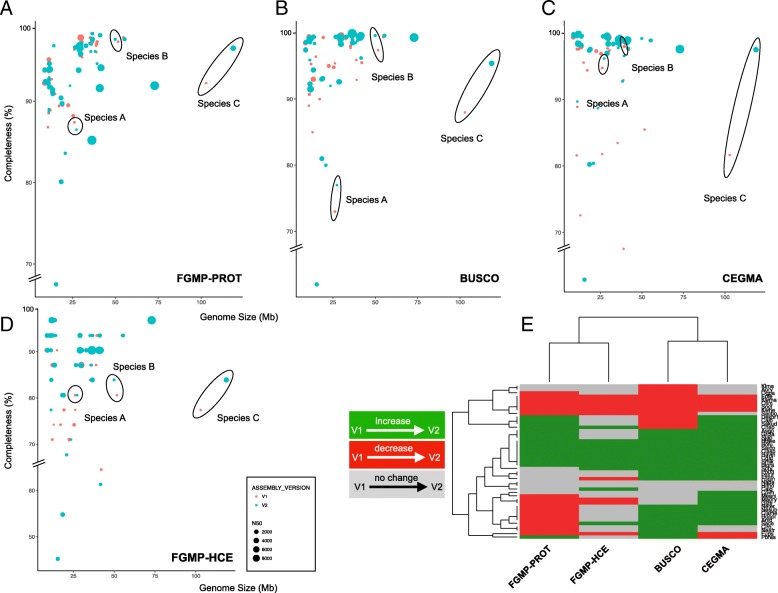
We propose a computational framework and sequence resource for assessing completeness of fungal genomes called FGMP (Fungal Genome Mapping Project). Our approach is based on evolutionary conserved sets of proteins and DNA elements and is applicable to various types of genomic data. We present a comparison of FGMP and state-of-the-art methods for genome completeness assessment utilizing 246 genome assemblies of fungi. We discuss genome assembly improvements/degradations in 57 cases where assemblies have been updated, as recorded by NCBI assembly archive.
FGMP is an accurate tool for quantifying level of completion from fungal genomic data. It is particularly useful for non-model organisms without reference genomes and can be used directly on unassembled reads, which can help reducing genome sequencing costs.
廉价的高通量 DNA 测序使大多数生物体的遗传信息民主化,因此利用生物体的基因组或转录组进行研究不再局限于模型系统。然而,采样基因组的组装质量差异很大,这阻碍了比较和有意义的解释。给定基因组序列完整性的不确定性可能限制了在许多谱系中报告的高基因丢失率模式的可行性。
我们提出了一种称为 FGMP(真菌基因组图谱绘制项目)的用于评估真菌基因组完整性的计算框架和序列资源。我们的方法基于进化保守的蛋白质和 DNA 元件集,适用于各种类型的基因组数据。我们利用 246 个真菌基因组组装进行了 FGMP 和最新基因组完整性评估方法的比较。我们讨论了在 57 个已更新组装的情况下的基因组组装改进/退化,这些情况都记录在 NCBI 组装档案中。
FGMP 是一种用于从真菌基因组数据中量化完成水平的准确工具。它对于没有参考基因组的非模式生物特别有用,并且可以直接用于未组装的读取,这有助于降低基因组测序成本。