Department of Environment and Sustainability, J. Craig Venter Institute, 4120 Capricorn Ln, La Jolla, CA, 92037, USA.
Department of Human Biology and Genomic Medicine, J. Craig Venter Institute, La Jolla, CA, 92037, USA.
BMC Bioinformatics. 2022 Oct 12;23(1):419. doi: 10.1186/s12859-022-04973-8.
With the advent of metagenomics, the importance of microorganisms and how their interactions are relevant to ecosystem resilience, sustainability, and human health has become evident. Cataloging and preserving biodiversity is paramount not only for the Earth's natural systems but also for discovering solutions to challenges that we face as a growing civilization. Metagenomics pertains to the in silico study of all microorganisms within an ecological community in situ, however, many software suites recover only prokaryotes and have limited to no support for viruses and eukaryotes.
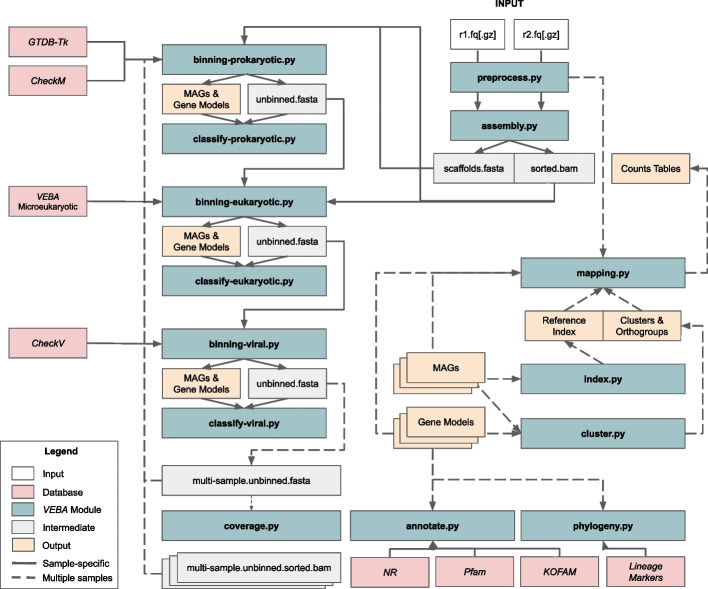
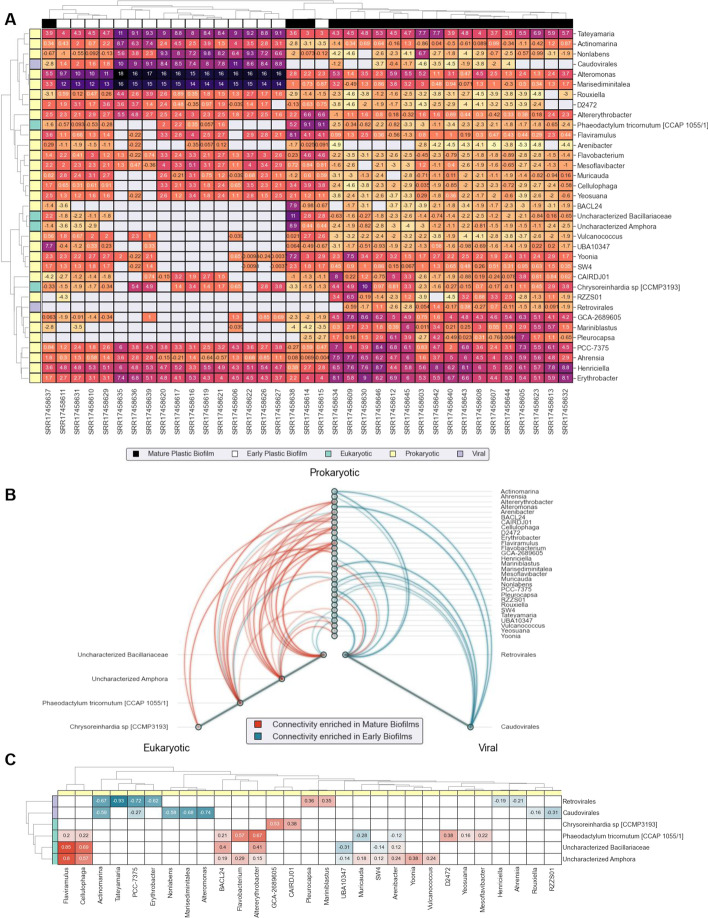
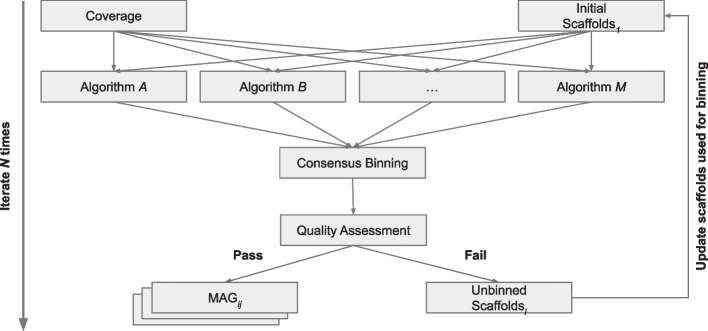
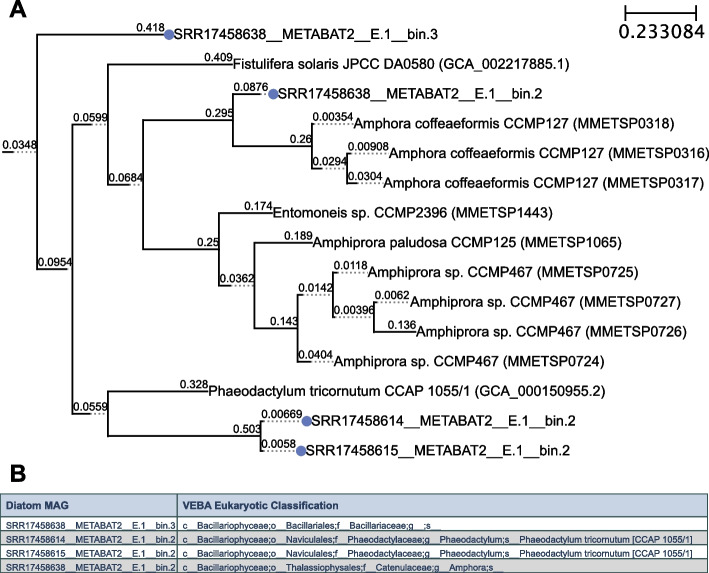
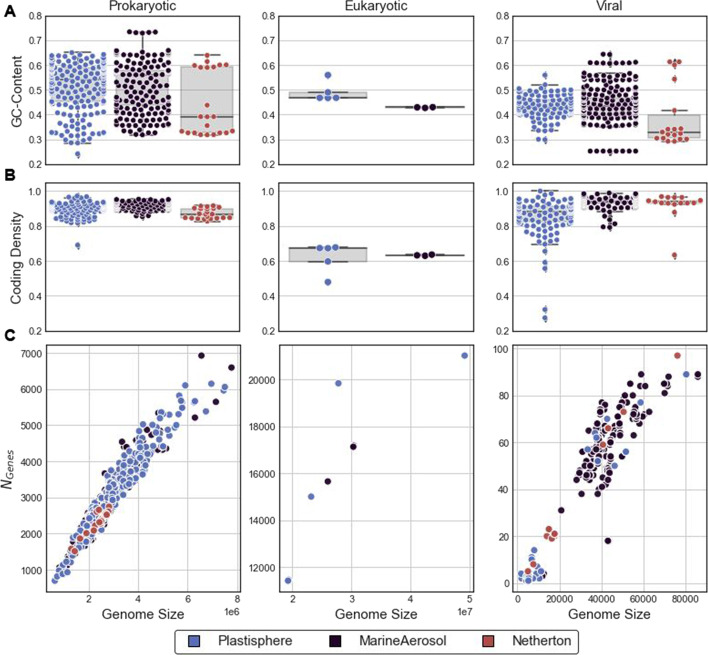
In this study, we introduce the Viral Eukaryotic Bacterial Archaeal (VEBA) open-source software suite developed to recover genomes from all domains. To our knowledge, VEBA is the first end-to-end metagenomics suite that can directly recover, quality assess, and classify prokaryotic, eukaryotic, and viral genomes from metagenomes. VEBA implements a novel iterative binning procedure and hybrid sample-specific/multi-sample framework that yields more genomes than any existing methodology alone. VEBA includes a consensus microeukaryotic database containing proteins from existing databases to optimize microeukaryotic gene modeling and taxonomic classification. VEBA also provides a unique clustering-based dereplication strategy allowing for sample-specific genomes and genes to be directly compared across non-overlapping biological samples. Finally, VEBA is the only pipeline that automates the detection of candidate phyla radiation bacteria and implements the appropriate genome quality assessments. VEBA's capabilities are demonstrated by reanalyzing 3 existing public datasets which recovered a total of 948 MAGs (458 prokaryotic, 8 eukaryotic, and 482 viral) including several uncharacterized organisms and organisms with no public genome representatives.
The VEBA software suite allows for the in silico recovery of microorganisms from all domains of life by integrating cutting edge algorithms in novel ways. VEBA fully integrates both end-to-end and task-specific metagenomic analysis in a modular architecture that minimizes dependencies and maximizes productivity. The contributions of VEBA to the metagenomics community includes seamless end-to-end metagenomics analysis but also provides users with the flexibility to perform specific analytical tasks. VEBA allows for the automation of several metagenomics steps and shows that new information can be recovered from existing datasets.
随着宏基因组学的出现,微生物及其相互作用与生态系统弹性、可持续性和人类健康的相关性的重要性变得显而易见。对生物多样性进行编目和保存不仅对地球的自然系统至关重要,而且对于发现我们作为一个不断发展的文明所面临的挑战的解决方案也至关重要。宏基因组学涉及对原位生态群落中所有微生物的计算机研究,然而,许多软件套件仅能恢复原核生物,并且对病毒和真核生物的支持有限或没有支持。
在这项研究中,我们介绍了病毒真核生物细菌古菌(VEBA)开源软件套件,该套件旨在从所有领域恢复基因组。据我们所知,VEBA 是第一个端到端的宏基因组学套件,可以直接从宏基因组中恢复、质量评估和分类原核生物、真核生物和病毒基因组。VEBA 实现了一种新颖的迭代分箱过程和混合样本特异性/多样本框架,比任何现有方法单独产生的基因组都多。VEBA 包含一个包含现有数据库中蛋白质的共识微真核生物数据库,以优化微真核生物基因建模和分类学分类。VEBA 还提供了一种独特的基于聚类的去重复策略,允许在非重叠的生物样本中直接比较特定于样本的基因组和基因。最后,VEBA 是唯一自动检测候选门辐射细菌并实施适当基因组质量评估的管道。VEBA 的功能通过重新分析 3 个现有的公共数据集得到了证明,共恢复了 948 个 MAG(458 个原核生物、8 个真核生物和 482 个病毒),包括几个未被表征的生物体和没有公共基因组代表的生物体。
VEBA 软件套件通过以新颖的方式整合前沿算法,允许从生命的所有领域的计算机中恢复微生物。VEBA 以最小化依赖性和最大化生产力的模块化架构完全集成了端到端和特定于任务的宏基因组分析。VEBA 为宏基因组学社区做出的贡献包括无缝的端到端宏基因组学分析,但也为用户提供了执行特定分析任务的灵活性。VEBA 允许自动化几个宏基因组学步骤,并表明可以从现有数据集中恢复新信息。