Department of Experimental Medicine, University of Campania "Luigi Vanvitelli", Via Luciano Armanni, 5, 80138 Napoli, Naples, Italy.
Department of Biomedical, Surgical and Dental Sciences, University of Milan, Milan, Italy.
J Exp Clin Cancer Res. 2019 Apr 12;38(1):160. doi: 10.1186/s13046-019-1164-5.
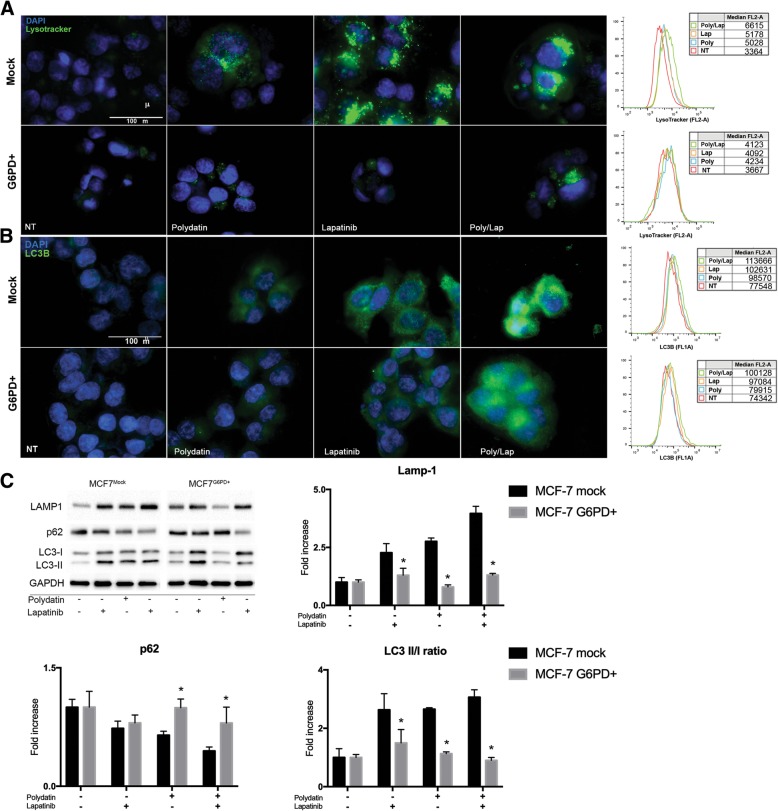
Glucose-6-phospate dehydrogenase (G6PD) is the limiting enzyme of the pentose phosphate pathway (PPP) correlated to cancer progression and drug resistance. We previously showed that G6PD inhibition leads to Endoplasmic Reticulum (ER) stress often associated to autophagy deregulation. The latter can be induced by target-based agents such as Lapatinib, an anti-HER2 tyrosine kinase inhibitor (TKI) largely used in breast cancer treatment.
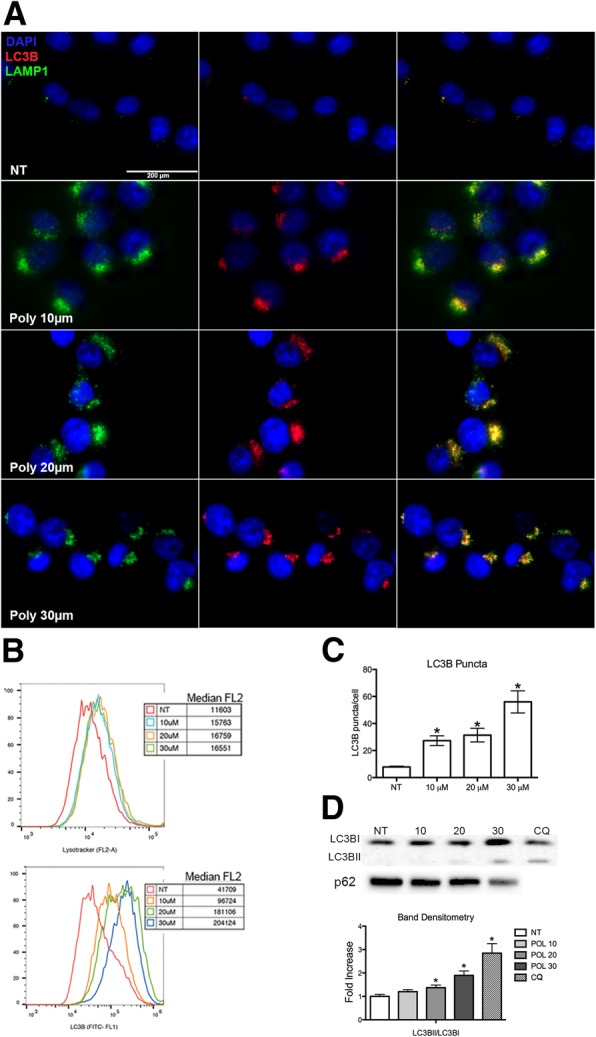
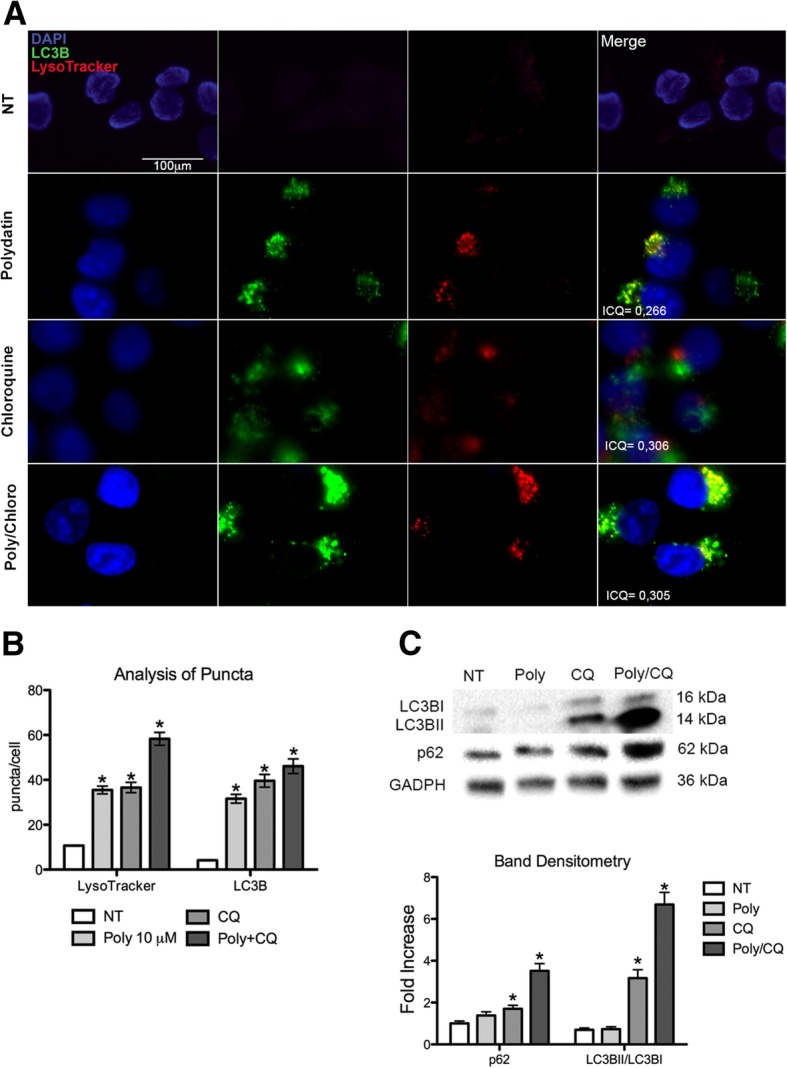
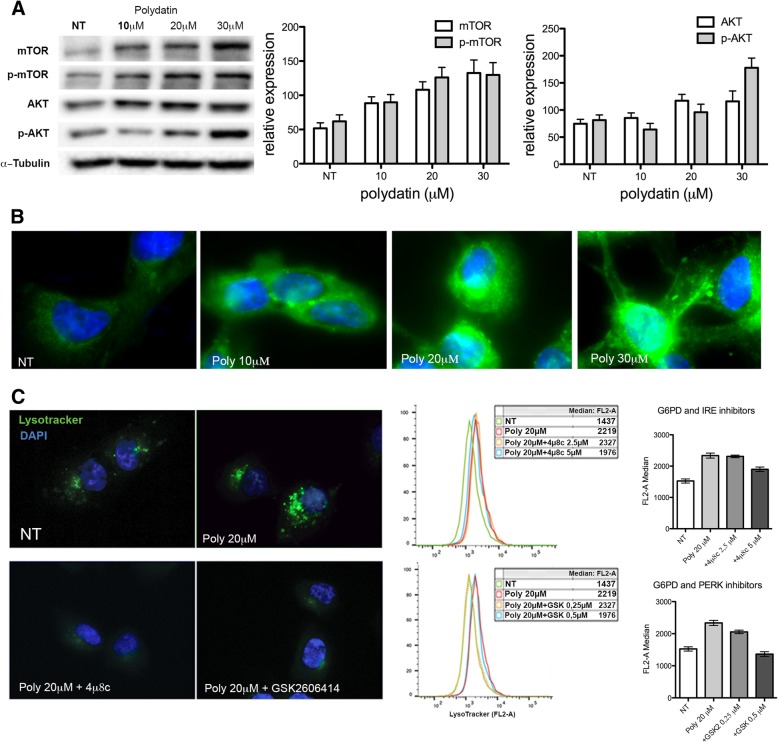
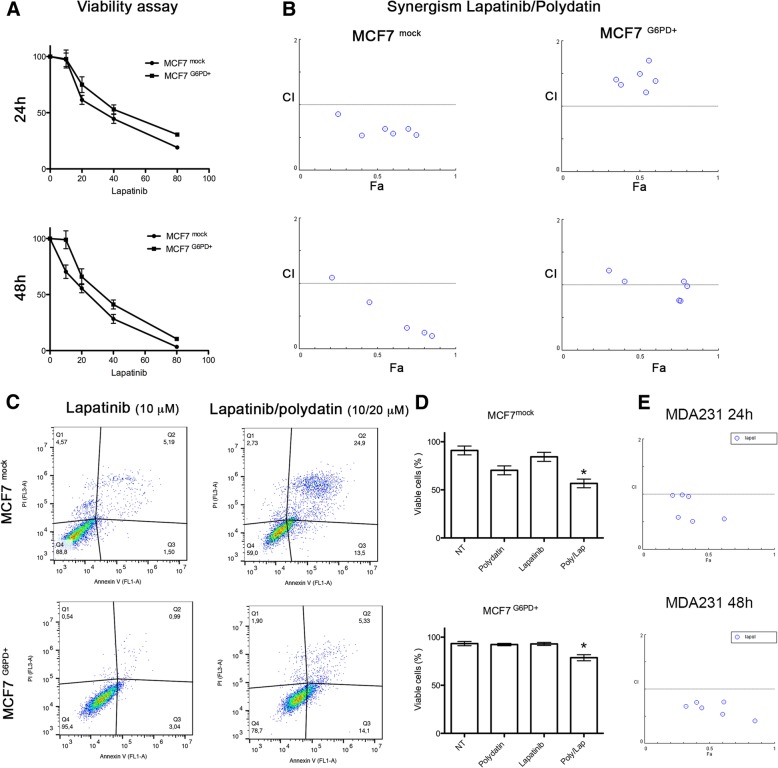
Here we investigate whether G6PD inhibition causes autophagy alteration, which can potentiate Lapatinib effect on cancer cells. Immunofluorescence and flow cytometry for LC3B and lysosomes tracker were used to study autophagy in cells treated with lapatinib and/or G6PD inhibitors (polydatin). Immunoblots for LC3B and p62 were performed to confirm autophagy flux analyses together with puncta and colocalization studies. We generated a cell line overexpressing G6PD and performed synergism studies on cell growth inhibition induced by Lapatinib and Polydatin using the median effect by Chou-Talay. Synergism studies were additionally validated with apoptosis analysis by annexin V/PI staining in the presence or absence of autophagy blockers.
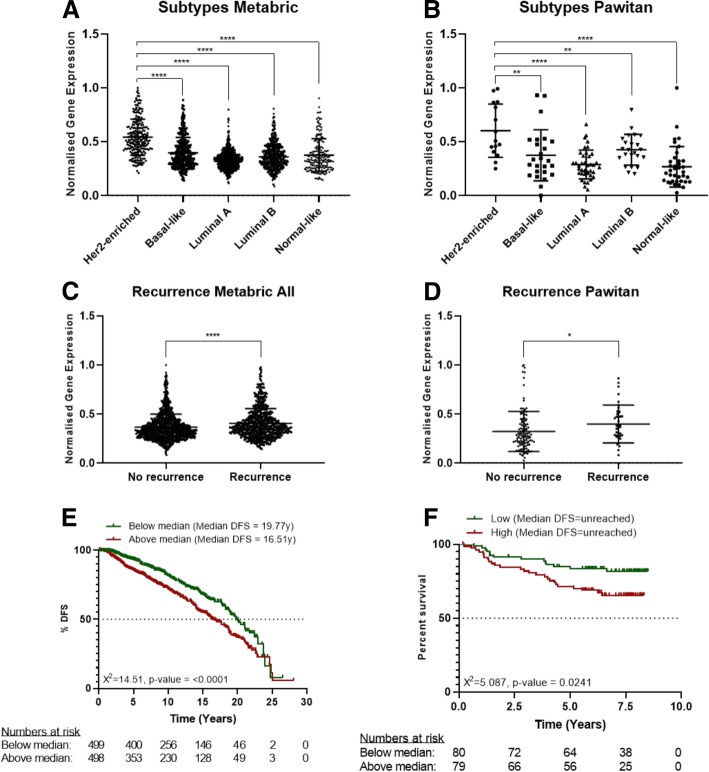
We found that the inhibition of G6PD induced endoplasmic reticulum stress, which was responsible for the deregulation of autophagy flux. Indeed, G6PD blockade caused a consistent increase of autophagosomes formation independently from mTOR status. Cells engineered to overexpress G6PD became resilient to autophagy and resistant to lapatinib. On the other hand, G6PD inhibition synergistically increased lapatinib-induced cytotoxic effect on cancer cells, while autophagy blockade abolished this effect. Finally, in silico studies showed a significant correlation between G6PD expression and tumour relapse/resistance in patients.
These results point out that autophagy and PPP are crucial players in TKI resistance, and highlight a peculiar vulnerability of breast cancer cells, where impairment of metabolic pathways and autophagy could be used to reinforce TKI efficacy in cancer treatment.
葡萄糖-6-磷酸脱氢酶(G6PD)是戊糖磷酸途径(PPP)的限速酶,与癌症进展和耐药性相关。我们之前的研究表明,G6PD 抑制会导致内质网(ER)应激,通常与自噬失调有关。后者可以被基于靶点的药物诱导,如拉帕替尼,一种广泛用于乳腺癌治疗的抗 HER2 酪氨酸激酶抑制剂(TKI)。
在这里,我们研究了 G6PD 抑制是否会导致自噬改变,从而增强拉帕替尼对癌细胞的作用。用 LC3B 和溶酶体示踪剂的免疫荧光和流式细胞术研究细胞中拉帕替尼和/或 G6PD 抑制剂(虎杖苷)处理后的自噬。用 LC3B 和 p62 的免疫印迹来验证自噬流分析,并进行点状和共定位研究。我们生成了一个过表达 G6PD 的细胞系,并使用 Chou-Talay 的中位数效应进行了拉帕替尼和虎杖苷诱导的细胞生长抑制的协同作用研究。协同作用研究还通过在存在或不存在自噬抑制剂的情况下用 Annexin V/PI 染色进行凋亡分析进行了验证。
我们发现,G6PD 的抑制诱导内质网应激,这是自噬流失调的原因。事实上,G6PD 阻断导致自噬体形成的一致增加,而与 mTOR 状态无关。过表达 G6PD 的细胞对自噬具有抗性,并对拉帕替尼产生耐药性。另一方面,G6PD 抑制协同增加了拉帕替尼对癌细胞的细胞毒性作用,而自噬阻断则消除了这种作用。最后,计算机模拟研究显示 G6PD 表达与患者肿瘤复发/耐药之间存在显著相关性。
这些结果表明,自噬和 PPP 是 TKI 耐药的关键因素,并强调了乳腺癌细胞的一种特殊脆弱性,其中代谢途径和自噬的损伤可以用于增强癌症治疗中 TKI 的疗效。