Bioinformatics Group, Department of Computer Science & Interdisciplinary Center for Bioinformatics, Universität Leipzig, Härtelstraße 16-18, Leipzig, 04107, Germany.
Max Planck Institute for Mathematics in the Sciences, Inselstraße 22, Leipzig, 04103, Germany.
BMC Bioinformatics. 2019 Apr 16;20(1):190. doi: 10.1186/s12859-019-2786-5.
The rapid increase in High-throughput sequencing of RNA (RNA-seq) has led to tremendous improvements in the detection and reconstruction of both expressed coding and non-coding RNA transcripts. Yet, the complete and accurate annotation of the complex transcriptional output of not only the human genome has remained elusive. One of the critical bottlenecks in this endeavor is the computational reconstruction of transcript structures, due to high noise levels, technological limits, and other biases in the raw data.
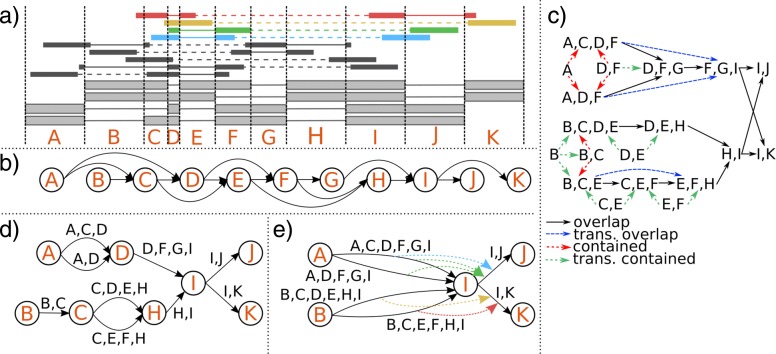
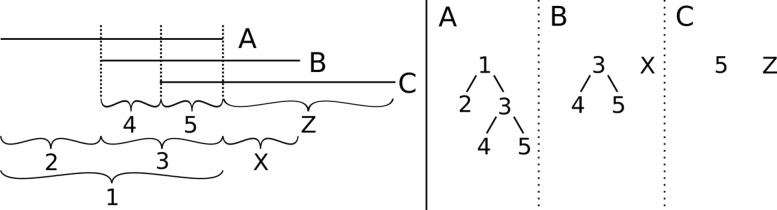
We introduce several new and improved algorithms in a novel workflow for transcript assembly and quantification. We propose an extension of the common splice graph framework that combines aspects of overlap and bin graphs and makes it possible to efficiently use both multi-splice and paired-end information to the fullest extent. Phasing information of reads is used to further resolve loci. The decomposition of read coverage patterns is modeled as a minimum-cost flow problem to account for the unavoidable non-uniformities of RNA-seq data.
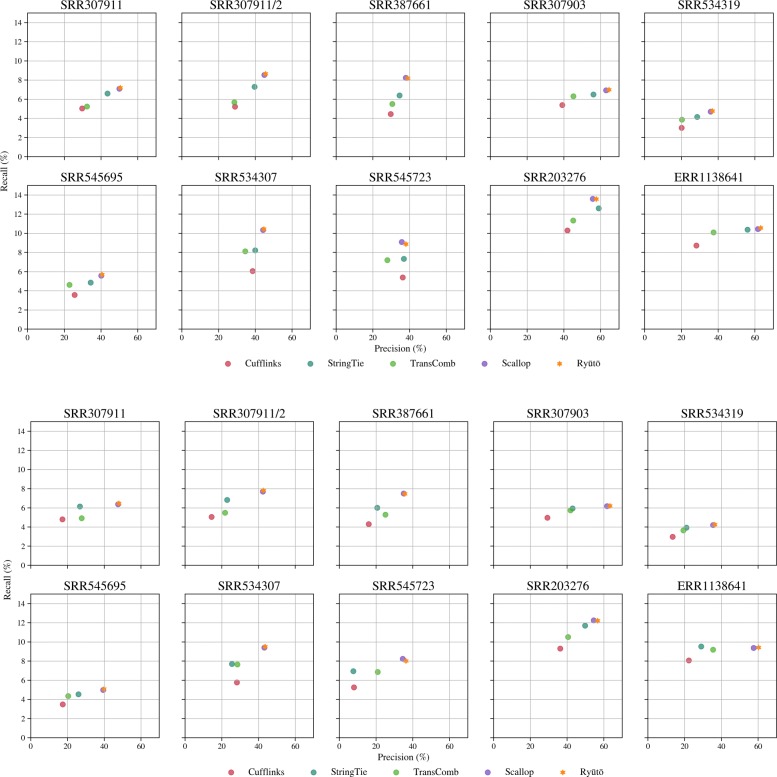
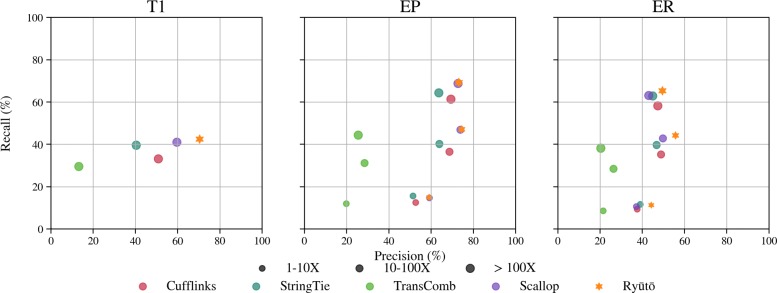
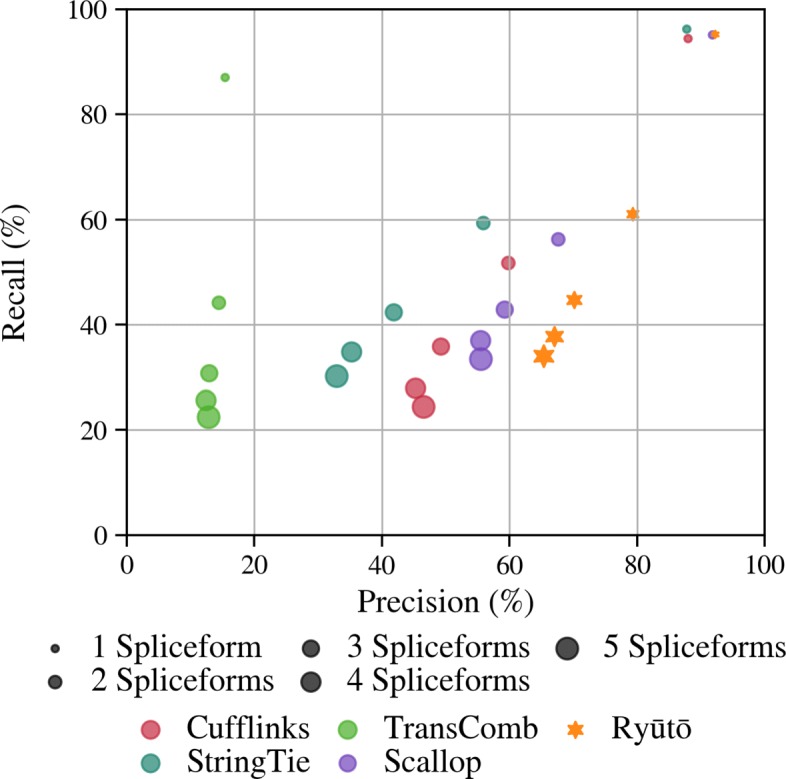
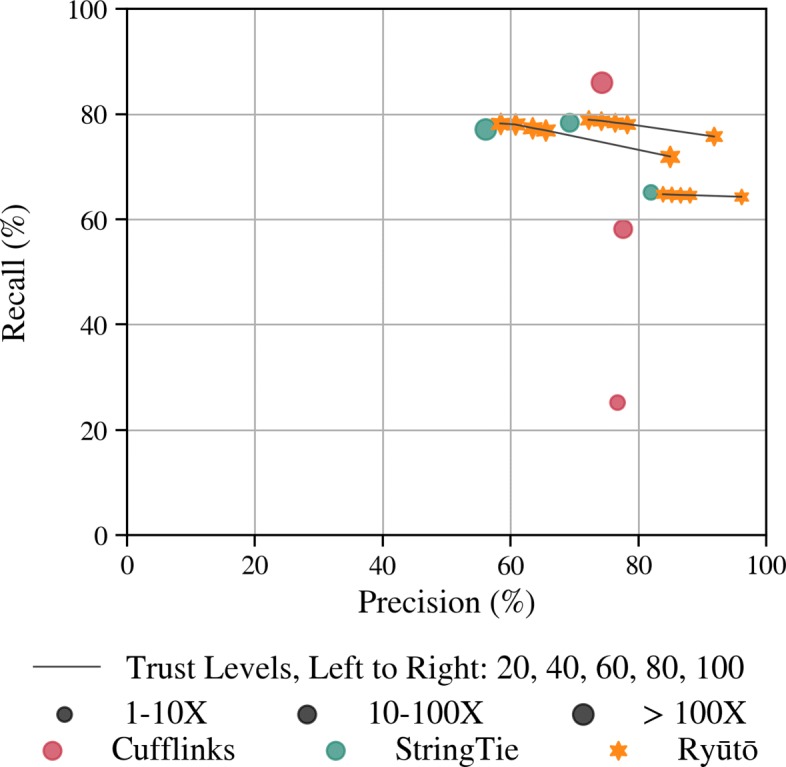
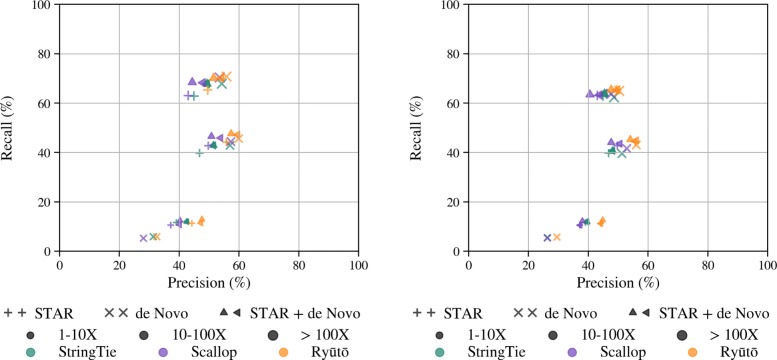
Its performance compares favorably with state of the art methods on both simulated and real-life datasets. Ryūtō calls 1-4% more true transcripts, while calling 5-35% less false predictions compared to the next best competitor.
高通量测序 RNA(RNA-seq)的快速发展,使得对表达编码和非编码 RNA 转录本的检测和重构都有了巨大的改进。然而,不仅人类基因组的复杂转录产物的完整和准确注释仍然难以实现。在这方面努力的一个关键瓶颈是由于高噪声水平、技术限制和原始数据中的其他偏差,转录本结构的计算重建。
我们在一个新的转录物组装和定量的工作流程中引入了几个新的和改进的算法。我们提出了一种常见剪接图框架的扩展,该框架结合了重叠和 bin 图的方面,使得能够充分利用多剪接和配对末端信息。读取的相位信息用于进一步解决基因座。读取覆盖模式的分解被建模为最小成本流问题,以解释 RNA-seq 数据不可避免的非均匀性。
它的性能在模拟和真实数据集上都优于最先进的方法。与下一个最佳竞争对手相比,Ryūtō 调用的真实转录本多 1-4%,而错误预测少 5-35%。