Institute of Chemical Biology and Fundamental Medicine, SBRAN, Novosibirsk, 630090, Russia.
Biosoft.ru, Ltd, Novosibirsk, 630090, Russia.
BMC Bioinformatics. 2019 Apr 18;20(Suppl 4):119. doi: 10.1186/s12859-019-2687-7.
The search for molecular biomarkers of early-onset colorectal cancer (CRC) is an important but still quite challenging and unsolved task. Detection of CpG methylation in human DNA obtained from blood or stool has been proposed as a promising approach to a noninvasive early diagnosis of CRC. Thousands of abnormally methylated CpG positions in CRC genomes are often located in non-coding parts of genes. Novel bioinformatic methods are thus urgently needed for multi-omics data analysis to reveal causative biomarkers with a potential driver role in early stages of cancer.
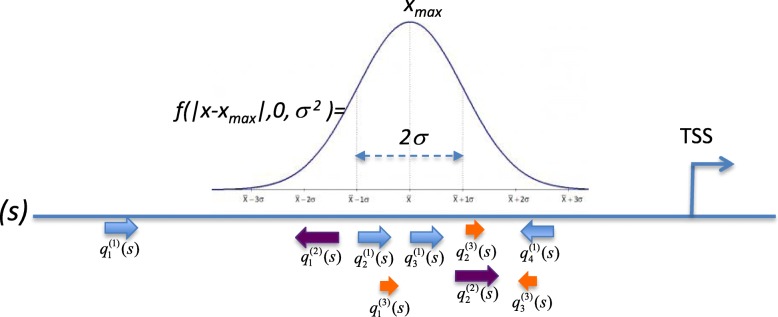
We have developed a method for finding potential causal relationships between epigenetic changes (DNA methylations) in gene regulatory regions that affect transcription factor binding sites (TFBS) and gene expression changes. This method also considers the topology of the involved signal transduction pathways and searches for positive feedback loops that may cause the carcinogenic aberrations in gene expression. We call this method "Walking pathways", since it searches for potential rewiring mechanisms in cancer pathways due to dynamic changes in the DNA methylation status of important gene regulatory regions ("epigenomic walking").
In this paper, we analysed an extensive collection of full genome gene-expression data (RNA-seq) and DNA methylation data of genomic CpG islands (using Illumina methylation arrays) generated from a sample of tumor and normal gut epithelial tissues of 300 patients with colorectal cancer (at different stages of the disease) (data generated in the EU-supported SysCol project). Identification of potential epigenetic biomarkers of DNA methylation was performed using the fully automatic multi-omics analysis web service "My Genome Enhancer" (MGE) (my-genome-enhancer.com). MGE uses the database on gene regulation TRANSFAC®, the signal transduction pathways database TRANSPATH®, and software that employs AI (artificial intelligence) methods for the analysis of cancer-specific enhancers.
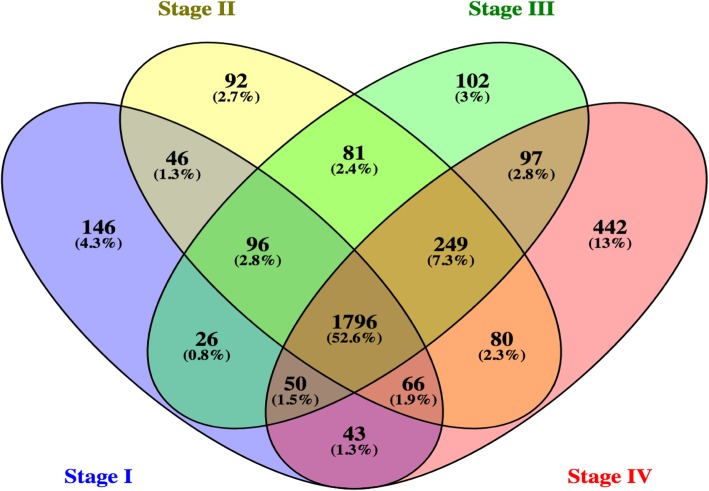
The identified biomarkers underwent experimental testing on an independent set of blood samples from patients with colorectal cancer. As a result, using advanced methods of statistics and machine learning, a minimum set of 6 biomarkers was selected, which together achieve the best cancer detection potential. The markers include hypermethylated positions in regulatory regions of the following genes: CALCA, ENO1, MYC, PDX1, TCF7, ZNF43.
寻找早发性结直肠癌(CRC)的分子生物标志物是一项重要但极具挑战性且尚未解决的任务。从血液或粪便中获取的人类 DNA 中的 CpG 甲基化检测已被提出作为一种非侵入性的 CRC 早期诊断的有前途的方法。CRC 基因组中数千个异常甲基化的 CpG 位置通常位于基因的非编码部分。因此,迫切需要新的生物信息学方法来进行多组学数据分析,以揭示具有癌症早期潜在驱动作用的因果生物标志物。
我们开发了一种方法,用于寻找影响转录因子结合位点(TFBS)的基因调控区域中的表观遗传变化(DNA 甲基化)与基因表达变化之间的潜在因果关系。该方法还考虑了所涉及的信号转导途径的拓扑结构,并搜索可能导致基因表达致癌异常的正反馈回路。我们将这种方法称为“行走途径”,因为它搜索由于重要基因调控区域的 DNA 甲基化状态的动态变化而导致癌症途径中潜在的重新布线机制(“表观基因组行走”)。
在本文中,我们分析了来自 300 名结直肠癌患者的肿瘤和正常肠道上皮组织样本的广泛的全基因组基因表达数据(RNA-seq)和基因组 CpG 岛的 DNA 甲基化数据(使用 Illumina 甲基化阵列)(在欧盟支持的 SysCol 项目中生成)。使用全自动多组学分析网络服务“我的基因组增强器”(MGE)(my-genome-enhancer.com)进行潜在表观遗传生物标志物的 DNA 甲基化鉴定。MGE 使用基因调控 TRANSFAC®数据库、信号转导途径数据库 TRANSPATH®以及采用人工智能(AI)方法分析癌症特异性增强子的软件。
鉴定的生物标志物在来自结直肠癌患者的独立血液样本集上进行了实验测试。结果,使用先进的统计和机器学习方法,选择了最小的 6 个生物标志物集合,它们共同实现了最佳的癌症检测潜力。这些标志物包括以下基因调控区域的超甲基化位置:CALCA、ENO1、MYC、PDX1、TCF7、ZNF43。