Department of Healthcare Policy and Research, Cornell University Weill Cornell, New York, New York 10065
Department of Biostatistics, University of North Carolina, Chapel Hill, North Carolina 27599.
Genetics. 2019 Jun;212(2):397-415. doi: 10.1534/genetics.119.301906. Epub 2019 Apr 22.
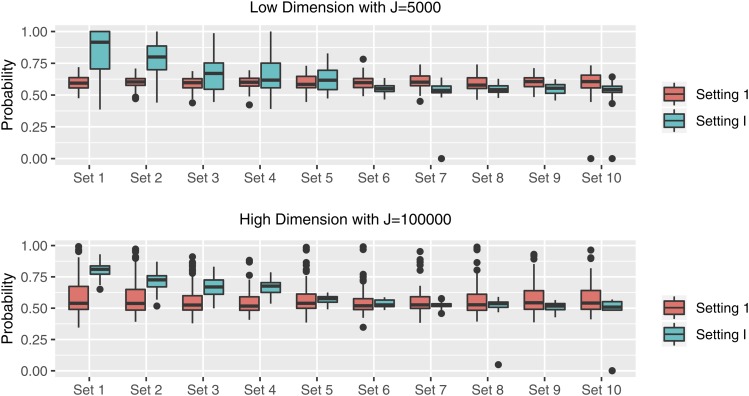
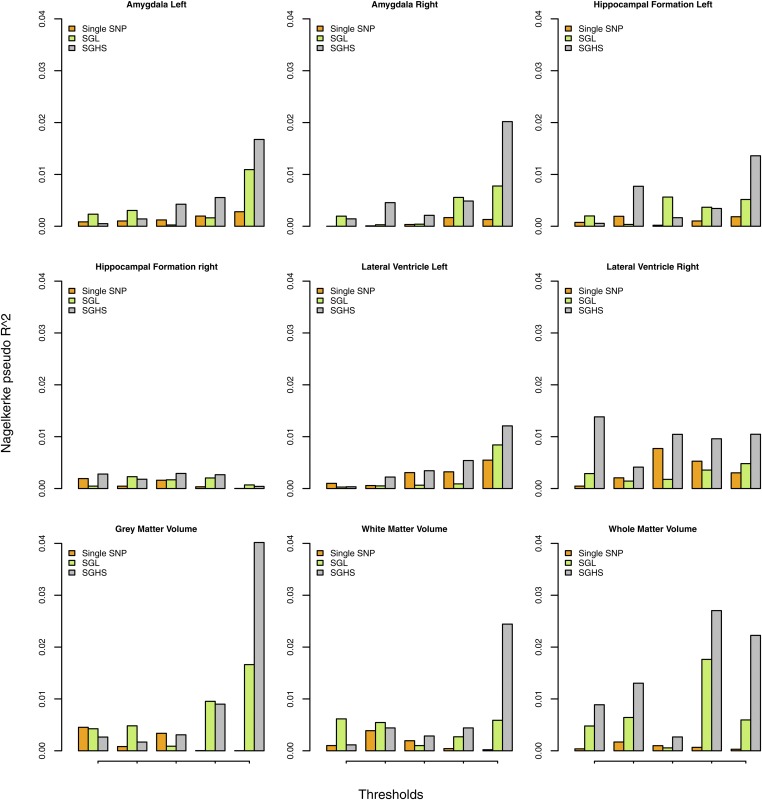
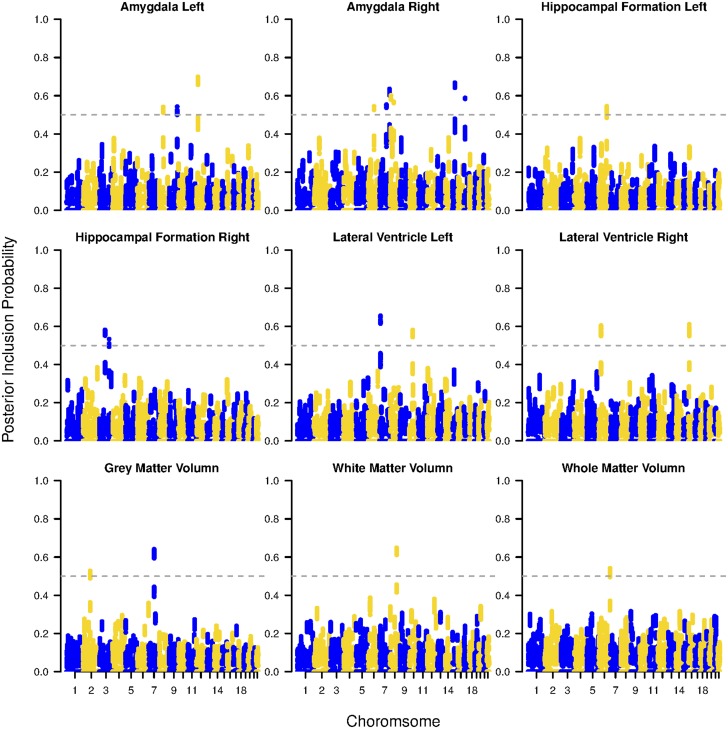
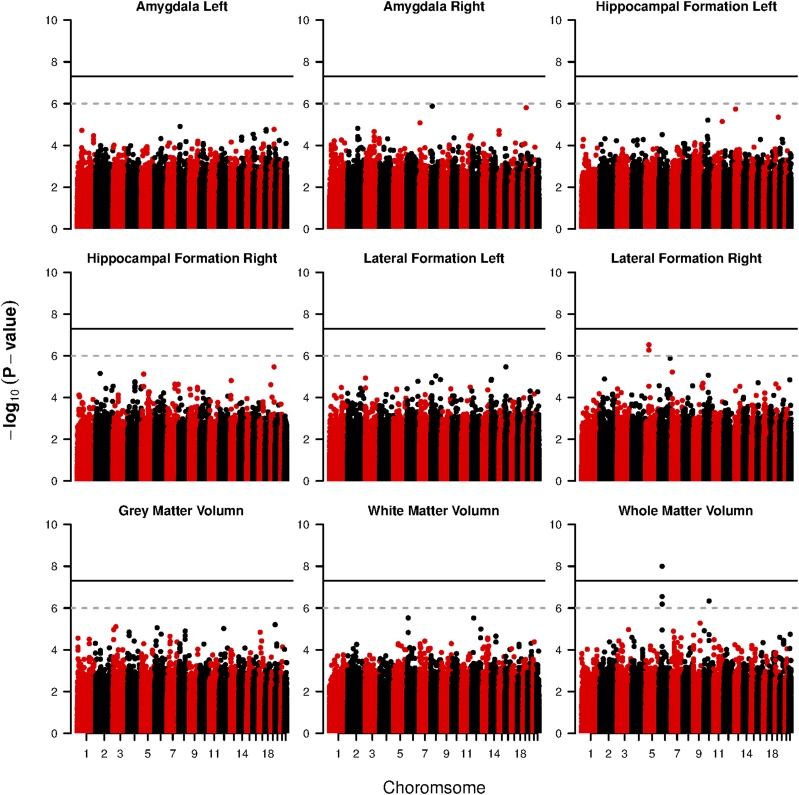
It becomes increasingly important in using genome-wide association studies (GWAS) to select important genetic information associated with qualitative or quantitative traits. Currently, the discovery of biological association among SNPs motivates various strategies to construct SNP-sets along the genome and to incorporate such set information into selection procedure for a higher selection power, while facilitating more biologically meaningful results. The aim of this paper is to propose a novel Bayesian framework for hierarchical variable selection at both SNP-set (group) level and SNP (within group) level. We overcome a key limitation of existing posterior updating scheme in most Bayesian variable selection methods by proposing a novel sampling scheme to explicitly accommodate the ultrahigh-dimensionality of genetic data. Specifically, by constructing an auxiliary variable selection model under SNP-set level, the new procedure utilizes the posterior samples of the auxiliary model to subsequently guide the posterior inference for the targeted hierarchical selection model. We apply the proposed method to a variety of simulation studies and show that our method is computationally efficient and achieves substantially better performance than competing approaches in both SNP-set and SNP selection. Applying the method to the Alzheimers Disease Neuroimaging Initiative (ADNI) data, we identify biologically meaningful genetic factors under several neuroimaging volumetric phenotypes. Our method is general and readily to be applied to a wide range of biomedical studies.
在使用全基因组关联研究(GWAS)来选择与定性或定量性状相关的重要遗传信息时,这一点变得越来越重要。目前,SNP 之间的生物关联的发现激发了各种策略来构建基因组上的 SNP 集,并将这种集合信息纳入选择过程中,以提高选择能力,同时促进更具生物学意义的结果。本文的目的是提出一种新的贝叶斯框架,用于 SNP 集(组)水平和 SNP(组内)水平的分层变量选择。我们通过提出一种新的抽样方案来克服大多数贝叶斯变量选择方法中现有后验更新方案的一个关键限制,该方案明确地适应了遗传数据的超高维性。具体来说,通过在 SNP 集水平下构建一个辅助变量选择模型,新的程序利用辅助模型的后验样本,随后指导目标分层选择模型的后验推断。我们将所提出的方法应用于各种模拟研究,并表明我们的方法在 SNP 集和 SNP 选择方面都具有计算效率,并且比竞争方法具有显著更好的性能。将该方法应用于阿尔茨海默病神经影像学倡议(ADNI)数据,我们在几种神经影像学体积表型下确定了具有生物学意义的遗传因素。我们的方法具有通用性,可广泛应用于各种生物医学研究。